Reductive cleavage of N-substituted aromatic amides as tert-butyl acylcarbamates†
Ulf
Ragnarsson
*a,
Leif
Grehn
a,
Hernani L. S.
Maia
b and
Luis S.
Monteiro
b
aDepartment of Biochemistry, University of Uppsala, Biomedical Center, PO Box 576, SE-751 23 Uppsala, Sweden. E-mail: urbki@bmc.uu.se; Fax: +46 18 55 21 39
bDepartamento de Quimica, Universidade do Minho, Gualtar, P-4700-320 Braga, Portugal
First published on 5th December 2001
Abstract
Synthetic and spectroscopic details relating to a set of heteroaromatic N-benzyl carboxamides and in particular the corresponding tert-butyl acylcarbamates are reported. These compounds were required to study the postulated effect of various heterocycles (pyridine and pyrazine with and without condensed benzene rings) on the cleavage of acyl–N bonds by reduction. All compounds were initially characterized by cyclic voltammetry (CV) which indicated various degrees of facilitated reduction, reflecting a direct influence of the heterocyclic component. Selected acylcarbamates were studied with respect to acyl–N bond cleavage by mild reducing agents, and selectively deacylated by activated aluminium and sodium borohydride. Conversion to acylcarbamates followed by reduction might therefore be a mild, efficient two-step procedure to effect cleavage of amides, allowing isolation of carbamates and with sodium borohydride also the corresponding alcohols.
Introduction
A very large number of methods and reagents are currently available for the synthesis of amides,1 a fact that reflects the prominence these compounds have reached in different areas. The high stability of the amide bond makes it hard to cleave, if desired, and requires aggressive chemicals, often not compatible with other reactive sites simultaneously present. Therefore, conversion to amides has only been used in a few cases for the protection of amino functions,2 for which carbamates in particular are currently preferred, since they can be tuned to cleave under mildly acidic, basic or other conditions.Electrochemical cleavage of protecting groups is occasionally feasible3 but only seldom practised. Among groups amenable to deprotection in this way is N-benzoyl, and on cathodic reduction benzamides provide the corresponding amines with concomitant formation of benzyl alcohol.4 A drawback with N-benzoyl is that rather negative potentials are required and therefore we are searching for more labile alternatives. To study the effect of structural changes on cathodic cleavage potentials, cyclic voltammetry (CV) is especially convenient and by using this technique we obtained evidence for facilitated electrochemical cleavage by introduction of a tert-butoxycarbonyl (Boc) group on the amide nitrogen.5 Parallel work was conducted on aromatic sulfonamides, which indicated a strong influence of the aromatic component on their activation potentials and allowed us to develop mild reductive cleavage and more labile sulfonamide protecting groups.6 This paper details related synthetic work dealing with carboxamides of the benzamide type, in which the phenyl ring has been replaced by naphthyl, pyridyl, pyrazyl, 2-quinolyl and quinoxalin-2-yl, as well as the corresponding N-Boc derivatives, together with typical reductive cleavage experiments. Since the corresponding heteroaromatic systems are progressively more easily reduced,7 our working hypothesis was that incorporation of such systems in conjugation with the amide bonds would facilitate reductive cleavage of the latter. A brief report with some key CV data and a summary of results demonstrating, for the first time, C(O)–N(Boc) bond cleavage by mild reducing agents has recently been published.8
Results
As a starting point for the project, a set of aromatic N-benzyl carboxamides (1), all previously described in the literature, and the corresponding N-benzyl N-Boc-arylcarboxamides or N-benzyl tert-butyl acylcarbamates (2), none of which had been described before, have been prepared and carefully characterized (Scheme 1).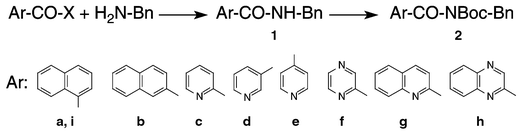 | ||
| Scheme 1 | ||
Most members of the subset 1 were prepared, as indicated in this scheme, from the acid chlorides (X = Cl), and then reacted to form 2 with Boc2O in the presence of 4-dimethylaminopyridine (DMAP) in order to introduce N-Boc groups onto their amide functions9a without additional tertiary amines, and with only catalytic amounts of DMAP (see typical procedure).9b The second step was monitored by TLC and was in some cases remarkably slow, requiring addition of extra Boc2O to go to completion, but ultimately afforded recrystallized, analytically pure products in yields of 75–91%.
 | ||
| Fig. 1 | ||
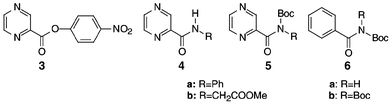
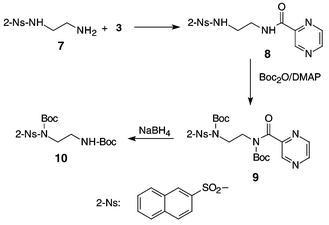
Additional compounds 4 and 5 (Fig. 1) relating to 1f and 2f with R = phenyl and methoxycarbonylmethyl have also been synthesized from inexpensive pyrazinecarboxylic acid. For the preparation of 4 we chose 3 as a starting material.10 This acylation agent, previously used to make a set of potential antituberculosis agents, was judged to be more practical than the corresponding unstable acid chloride. The dicyclohexylcarbodiimide-mediated condensation of pyrazinecarboxylic acid with 4-nitrophenol gave only 35% of 3 with significant concomitant formation of contaminating N-acylurea. The above acid was however, smoothly converted to 3 in 73% yield by bis(4-nitrophenyl) sulfite in dry pyridine.11 Compound 3 was reacted with 7 to give 8 and 9 (Scheme 2), which was used to demonstrate selective cleavage.
 | ||
| Scheme 2 | ||
All compounds 1, 2, 4–6 and 7–10 were studied by CV, of which the data for 1a–g and 2a–g were included in our preliminary communication.8 To summarize these data, the activation potentials of the pyridinecarboxamide and naphthamide derivatives were less negative than those of the corresponding benzamides, and the analogous pyrazine and quinoline compounds gave peaks at even higher values. As shown in Table 1 the values for 1h and 2h are −1.25 and −1.01 V which means that they are the highest in the two series of compounds studied so far. For a typical CV curve, see Fig. 2.
| Entry | Cmpd | Comments, R | −EP/Va |
|---|---|---|---|
| a Versus SCE; cathode, vitreous carbon; solvent, DMF; supporting electrolyte, Bu4NBF4 0.1 mol dm−3; substrate concentration, ∼0.005 mol dm−3. b N-Phenyl-1-naphthamide. c N-Boc-N-phenyl-1-naphthamide. | |||
| 1 | 1, 2a–g | See Table in ref. 8 | |
| 2 | 1h | 1.25 | |
| 3 | 2h | 1.01 | |
| 4 | 1i | Ar = 1-Naphthyl Ph instead of Bnb | 2.13 |
| 5 | 2i | Ar = 1-Naphthyl Ph instead of Bnc | 1.79 |
| 6 | 4a | Ph | 1.29 |
| 7 | 4b | CH2COOMe | 1.41 |
| 8 | 5a | Ph | 1.13 |
| 9 | 5b | CH2COOMe | 1.25 |
| 10 | 6a | H | 1.75 |
| 11 | 6b | Boc | 1.56 |
| 12 | 7 | 2.07 | |
| 13 | 8 | 1.75/∼2.3 | |
| 14 | 9 | 1.45/1.76 | |
| 15 | 10 | 1.79 | |
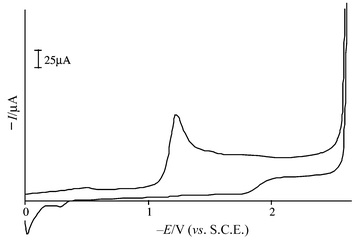 | ||
| Fig. 2 Cyclic voltammogram at a vitreous carbon electrode of 0.005 mol dm−3 solution of 2g in DMF, with 0.1 mol dm−3 Bu4NBF4 as supporting electrolyte, at a sweep rate of 100 mV s−1 (SCE = standard calomel electrode). | ||
tert-Butyl acylcarbamates are known to undergo fast aminolysis and alkaline hydrolysis with formation of Boc-amine,9a,c and in model experiments it has also been demonstrated that benzoylcarbamates can be cleaved selectively in the presence of benzamides by electrolysis.5b When compounds 2f and 2g were subjected to controlled potential electrolysis close to their modestly negative CV activation potentials, Boc-benzylamine could be isolated in 66 and 96% yield,8 demonstrating the significance of the CV data.
A large number of further reductive cleavage experiments have been carried out with 2 using activated aluminium in moist diethyl ether.12 This mild, essentially neutral system was recently shown to cleave S–N bonds in related sulfonamides with activation potentials above −1.7 to −1.8 V, 6c and compounds 2c–g were also cleaved under these conditions with formation of Boc–NH–Bn, directly isolated in yields of 89, 74–85, 90, 49 and 95%, respectively.8 On the other hand, the naphthoyl derivatives 2a, 2b and 2i underwent only partial cleavage of the acyl–N bonds and competing naphthalene reduction took place, requiring purification of these reaction mixtures by chromatography which allowed isolation of Boc–NH–Bn in 54–80% yield.
To gain additional information about the applicability of this seldom used reduction method, a few experiments have been performed in which two substrates were simultaneously reduced. By quenching these reactions before they had gone to completion and determining the ratio of the remaining starting materials, some information about their relative tendency to undergo reaction with activated aluminium could be obtained. These experiments were performed with 2c, 2d, 2f and 2g, using the first compound as a reference, and the data, shown in Table 2, were generated by integration of the appropriate Boc-Me and/or benzyl-CH2 signals from 1H NMR spectra of samples derived by evaporation of the solvent directly from the reduction experiments. The results indicate that 2d is reduced relatively slowly, whereas 2c reacts faster, faster also than 2g with significantly less negative activation potential. With respect to the low yield in the preparative cleavage of 2f, the data under entry 3 may be less reliable.
| Entry | Compds | Reagent ratioa | Remaining 2x/2cb | Relative rate | Conversion ratioc |
|---|---|---|---|---|---|
| a Molecular ratio 2x ∶ 2c ∶ activated aluminium. b By integration of the appropriate Boc-Me and/or Bn-CH2 signals of 1H NMR spectra. c Ratio between Boc–NHBn and total remaining starting material by integration of these signals. | |||||
| 1 | 2d, 2c | 1 ∶ 1 ∶ 2 | 1.3 | 2c ≥ 2d | 0.8 |
| 2 | 2d, 2c | 1 ∶ 1 ∶ 6 | >10 | 2c > 2d | 40 |
| 3 | 2f, 2c | 1 ∶ 1 ∶ 2 | 0.7 | 2f > 2c? | 0.7 |
| 4 | 2g, 2c | 1 ∶ 1 ∶ 2 | 1.2 | 2c ≥ 2g | 0.2 |
| 5 | 2g, 2c | 1 ∶ 1 ∶ 6 | 3.4 | 2c > 2g | 9.0 |
Among other, more conventional reducing agents with potential application in the cleavage of acylcarbamates, significant results have been obtained with NaBH4. This convenient, extremely mild reagent was introduced by Weygand and Frauendorfer for the removal of N-trifluoroacetyl and trichloroacetyl groups.13 Ganem et al. have developed a useful cleavage method for phthalimides based on partial reduction with this agent.14 The first qualitative experiment with NaBH4 in absolute ethanol at room temperature was undertaken with 2f, and TLC indicated not only a surprisingly fast but also an absolutely clean and complete conversion. These preliminary results could easily be confirmed in a preparative experiment (see typical experiment in the Experimental) in which pure Boc-benzylamine was obtained in 97% isolated yield. The pyridine derivative 2c gave similar results (98%, no experimental details given). It is also worth mentioning that the benzoyl derivative 6b with two Boc-groups on nitrogen is also cleaved by NaBH4.
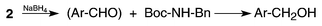 | ||
| Scheme 3 | ||
The initial step in the reduction of acylcarbamates with NaBH4 obviously involves a hydride transfer and should therefore lead to the formation of an aldehyde (Scheme 3), but since such compounds are easily and selectively converted to alcohols by this reagent, further reduction is likely to take place. This was proved in the case of 2a which upon chromatography of the crude reaction mixture on a silica column (see Supporting Information†) furnished pure 1-naphthylmethanol in an isolated yield of 90% in addition to 91% of the theoretical amount of Boc-benzylamine. However, careful monitoring by TLC indicated that the reduction of 2a took place more slowly than for 2c, requiring only a few hours, and particularly in comparison with 2f. Initially this seemed reasonable but it should be pointed out that 2g reacts significantly more slowly than 2f and therefore careful monitoring of this type of deacylation is strongly recommended.
At this stage we decided to prepare the ethylenediamine derivative 9 and subject it to cleavage with NaBH4 as described in the preceding paragraph. Again a very fast cleavage of the pyrazinecarbonyl group took place giving 10 but more importantly the otherwise comparatively labile naphthalene-2-sulfonyl (2-Ns) function6c was not at all affected (see Supporting Information†).Compounds 8–10 were all carefully characterized and additional CV data are shown in Table 1.
Discussion
An ideal protecting group should fulfil a number of criteria with respect to its introduction, stability to various reagents and final cleavage. Judged in this perspective, N-benzoyl obviously fails, since it cannot be selectively cleaved under mild conditions. Among derivatives of N-substituted benzamides or N-substituted amides in general, tert-butyl acylcarbamates are of particular interest, because they can be more easily cleaved by hydrolysis,9a methanolysis,9a aminolysis9c and electrolysis.6 Under the conditions used for formation of N-substituted tert-butyl acylcarbamates, N-unsubstituted amides give rise to di-tert-butyl acylimidodicarbonates.15a–e Selective mono-Boc protection was reported to be unsuccessful,16a but tert-butyl acylcarbamates have been prepared by RuO4-oxidation of carbamates16a–c and are stable compounds with increased sensitivity to acid and base in comparison with the corresponding nonacylated carbamates and amides.16d Di-tert-butyl benzoylimidodicarbonate 15b (N,N-bis-Boc-benzamide) was first shown to be an N-benzoylating agent and subsequently several other acylimidodicarbonates have been used for N-acylation.15b–eThe structures of two tert-butyl acylcarbamates including the benzoyl-N-phenyl species have previously been determined.17 In this molecule the benzoyl C(O)–N distance is increased by 0.058 Å in comparison with that of benzanilide, and the corresponding carbamate bond length by over 0.06 Å compared with typical Boc-amino acids. The shifts to less negative activation potentials observed on conversion of various model benzamides to benzoylcarbamates5 should also be highlighted at this point. Obviously tert-butoxycarbonylation profoundly affects the length and strength of the key amide bond by interaction of the two N–C(O) orbitals, resulting in a significant change in resonance stabilization of this bond and leading to its weakening.
As demonstrated with a set of N-benzyl derivatives,8 on substitution with a more readily reducible aromatic or heteroaromatic system than phenyl, the CV activation potentials of the corresponding amides and acylcarbamates are significantly shifted in comparison with the benzamides and benzoylcarbamates. In this paper various attempts have been made to characterize the stability of acyl–N bonds in acylcarbamates with respect to reductive cleavage. As already mentioned, 2f and 2g could be deacylated by controlled potential electrolysis above −1.5 V, i.e. at much less negative potential than that required to cleave benzoyl.
Selective cleavage of all acylcarbamates investigated with activation potentials above −1.75 V was accomplished with activated aluminium in moist diethyl ether with isolated yields around 90% except for 2f which seemed to give rise to a side-reaction. Attempts to correlate the rate of cleavage by aluminium within this group of substances with their CV activation potentials under competitive conditions were inconclusive.
Very clean and complete reductive cleavage of 2 in all compounds tested (a,c,f,g) was accomplished by NaBH4 in ethanol, allowing quantitative isolation of Boc–NHBn. Since this reagent attacks aldehyde and ketone carbonyls with formation of the corresponding alcohols essentially selectively, a simultaneous formation of such was anticipated and also demonstrated in one case (for 2a, see Supporting Information†). The simple conversion of amides to acylcarbamates and their easy and complete reduction by NaBH4 to alcohols might also occasionally be of preparative value.
Sulfonamides can be cleaved by reduction with various metals and metal salts12 but apart from that they are very stable to reducing agents including those based on hydride transfer. Previously we have desulfonylated sulfonylcarbamates by either controlled potential electrolysis or magnesium in anhydrous methanol.6a,b After it was found that acylcarbamates could be deacylated with NaBH4, as mentioned above, we prepared 9 and subjected it to reduction with this agent, whereupon a fast deacylation but no detectable desulfonylation took place. This indicates that ordinary sulfonylcarbamates are also stable to hydride transfer.
In summary, several N-acylcarbamates with acyl functions of the heteroaroyl type have been prepared and initially examined by cyclic voltammetry with respect to reduction. Of the compounds studied, those based on pyrazine, 2-quinoline and 2-quinoxaline in particular indicated highly facilitated reduction. Selected products were cleaved by controlled potential electrolysis above −1.5 V versus SCE and especially by the mild reducing agents, activated aluminium and NaBH4.
Experimental
General methods
All mps were measured on a Gallenkamp apparatus and are uncorrected. TLC analyses were carried out on 0.25 mm thick precoated silica plates (Merck Fertigplatten Kieselgel 60F254) and spots were visualized under UV light. FT-IR spectra were recorded for KBr disks at a resolution of 4 cm−1 on a Mattson Polaris spectrometer equipped with software for conversion to % transmission. 1H and 13C NMR spectra were recorded with a JEOL JMN Ex 400 spectrometer in ∼5% solution at 25 °C. All shifts are given in δ ppm using δH(TMS) = 0 and δC(CDCl3/DMSO-d6) = 77.02/39.50, respectively, as internal references. Elemental analyses were performed by Mikro Kemi AB (Uppsala).Synthesis of N-benzyl-N-Boc-arenecarboxamides (2)
Reductive cleavage experiments
Cleavage of 2g. Typical procedure. A two compartment cell for controlled potential electrolyses was filled with MeCN containing Et4NCl (0.1 mol dm−3) as the supporting electrolyte and Et3NHCl (0.015 mol dm−3) as the proton donor. Recrystallized 2g (253 mg, 0.65 mmol) was added to the cathodic compartment and a cyclic voltammogram was recorded at a sweep rate of 100 mV s−1 in order to measure the corresponding peak potential. The potential was adjusted to a value corresponding to 50 mV lower than the peak potential measured, and the apparatus switched on. When the intensity of the current was almost zero, the reaction mixture (catholyte) was transferred to a round-bottomed flask and the solvent evaporated at reduced pressure. The residue was partitioned between 100 cm3 of EtOAc and 50 cm3 of KHSO4 (1 mol dm−3). The organic phase was then washed with KHSO4 (1 mol dm−3), NaHCO3 (1 mol dm−3) and brine (3 × 30 cm3 each) and dried (MgSO4 in the presence of decolorizing carbon). Removal of the solvent left a slightly greenish oil. The yield of crude, essentially pure Boc–NHBzl was 129.4 mg (96%). Crystallization from n-hexane (decolorizing carbon) afforded the analytical specimen as white crystals; mp 54.0–55.0 °C. TLC (toluene–MeCN 2 ∶ 1) and 1H NMR showed complete identity with an authentic sample.
Activated Al foil. Aluminium foil (kitchen foil, ∼0.02 mm) was cut into small pieces (1–2 cm2) and carefully washed with light petroleum and Et2O. It was then covered with 0.5 mol dm−3 NaOH for about 30 s, whereupon small bubbles of gas appeared. The solution was quickly decanted and the foil was rapidly rinsed three times with glass-distilled water. A 0.5% solution of HgCl2 was added and left for 40–50 s, after which time the foil had attained a dark grey colour. The solution was decanted and the foil was quickly rinsed, in turn, with glass-distilled water, EtOH and finally with Et2O (3 times each) and then kept under Et2O.
Cleavage of compound 2e. Typical procedure. Recrystallized 2e (312 mg, 1.00 mmol) was dissolved in Et2O (35 cm3) containing water (1%) and treated with freshly activated Al foil (270 mg, 10 mmol) in small portions in a CO2(g) atmosphere with rapid stirring at RT. After 3 h, TLC (toluene–MeCN 2 ∶ 1) indicated complete consumption of 2e and the grayish solid was filtered off and rinsed repeatedly with Et2O. The combined filtrate and washings (30 cm3) were washed successively with 1 mol dm−3 KHSO4, 1 mol dm−3 NaHCO3 and brine (3 × 10 cm3 each) and dried (MgSO4 in the presence of decolorizing carbon). A white solid weighing 187 mg (90%) was obtained after meticulous drying at reduced pressure. TLC (Et2O–light petroleum 1 ∶ 2) and 1H NMR showed complete identity with an authentic sample.
Cleavage of compounds 2 by activated aluminium under competition conditions. General procedure. The two substrates (0.10 mmol each) were dissolved in Et2O containing 1% water (∼5 cm3) at RT under CO2. To the clear solution was added freshly activated Al [1 equiv. (∼6 mg) or 3 equiv. (∼17 mg)] with rapid stirring. After 4–6 h, most of the Al had reacted and the insoluble fine-grained solid was filtered off. The essentially colourless filtrate was evaporated to complete dryness and the oily residue was examined by 1H NMR (CDCl3). Integration was carried out over Boc-Me and/or benzyl-CH2 signals and molar ratios of the remaining substrates as well as those of products/starting materials were determined.
(a) Typical procedure with isolation only of Boc–NH–Bn. Compound 2f (313 mg, 1.00 mmol) in absolute EtOH (8 cm3) was treated with NaBH4 (76 mg, 2.00 mmol) with initial mechanical vibration or rapid stirring. The mixture became turbid within 10 min, and in this case the reaction was over after 10–15 min (monitoring by TLC). After 1 h the reaction was quenched by addition of acetone (1 cm3) and left for another hour, whereupon the mixture was taken to dryness and the remaining semisolid material partitioned between Et2O and 1 mol dm−3 KHSO4. The organic phase was washed successively with 1 mol dm−3 KHSO4, 1 mol dm−3 NaHCO3 and brine (3 times each) and dried (MgSO4). Evaporation gave an oil, pure by TLC, which slowly solidified on drying in vacuo; yield 97%. Recrystallization furnished glittering flakes, mp 56–56.5 °C, in close agreement with most literature values (48–58 °C).
(b) Reduction of compound 2a with isolation also of naphthalene-1-methanol. This experiment is described in the Supporting information.†
Acknowledgements
U. R. thanks NFR and Magnus Bergvalls Stiftelse for their generous support and the RSC for a journals grant for international authors. L. G. acknowledges a grant from TFR. We wish to acknowledge also the Foundation for Science and Technology (Portugal) for financial support to the Institute of Biotechnology and Fine Chemistry (University of Minho).References
- B. R. Brown, The Organic Chemistry of Aliphatic Nitrogen Compounds, Clarendon Press, Oxford, 1994, ch. 8 Search PubMed.
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 3rd edn., 1999, ch. 7 Search PubMed; (b) P. J. Kocieński, Protecting Groups, Thieme, Stuttgart, 1994, ch. 6. Search PubMed.
- Reviews: (a) V. G. Mairanovsky, Angew. Chem., 1976, 88, 283 (Angew. Chem., Int. Ed. Engl., 1976, 15, 281) Search PubMed; (b) M. I. Montenegro, Electrochim. Acta, 1986, 31, 607 CrossRef CAS.
- L. Horner and H. Neumann, Chem. Ber., 1965, 98, 3462 Search PubMed.
- (a) L. Grehn, K. Gunnarsson, H. L. S. Maia, M. I. Montenegro, L. Pedro and U. Ragnarsson, J. Chem. Res., 1988, (S) 399 Search PubMed; L. Grehn, K. Gunnarsson, H. L. S. Maia, M. I. Montenegro, L. Pedro and U. Ragnarsson, J. Chem. Res., 1988, (M) 3081 Search PubMed; (b) L. Grehn, H. L. S. Maia, L. S. Monteiro, M. I. Montenegro and U. Ragnarsson, J. Chem. Res., 1991, (S) 144 Search PubMed; L. Grehn, H. L. S. Maia, L. S. Monteiro, M. I. Montenegro and U. Ragnarsson, J. Chem. Res., 1991, (M) 1501 Search PubMed; (c) H. L. S. Maia, L. S. Monteiro, F. Degerbeck, L. Grehn and U. Ragnarsson, J. Chem. Soc., Perkin Trans. 2, 1993, 495 RSC.
- (a) B. Nyasse, L. Grehn, U. Ragnarsson, H. L. S. Maia, L. S. Monteiro, I. Leito, I. Koppel and J. Koppel, J. Chem. Soc., Perkin Trans. 1, 1995, 2025 RSC; (b) B. Nyasse, L. Grehn and U. Ragnarsson, Chem. Commun., 1997, 1017 RSC; (c) B. Nyasse, L. Grehn, H. L. S. Maia, L. S. Monteiro and U. Ragnarsson, J. Org. Chem., 1999, 64, 7135 CrossRef CAS.
- (a) B. J. Tabner and J. R. Yandle, J. Chem. Soc. A, 1968, 381 RSC; (b) K. B. Wiberg and T. P. Lewis, J. Am. Chem. Soc., 1970, 92, 7154 CrossRef CAS.
- U. Ragnarsson, L. Grehn, H. L. S. Maia and L. S. Monteiro, Org. Lett., 2001, 3, 2021 CrossRef CAS.
- (a) D. L. Flynn, R. E. Zelle and P. A. Grieco, J. Org. Chem., 1983, 48, 2424 CrossRef CAS; (b) L. Grehn, K. Gunnarsson and U. Ragnarsson, Acta Chem. Scand., Ser. B, 1986, 40, 745 Search PubMed; (c) L. Grehn, K. Gunnarsson and U. Ragnarsson, J. Chem. Soc., Chem. Commun., 1985, 1317 RSC.
- M. H. Cynamon, S. P. Klemens, T.-S. Chou, R. H. Gimi and J. T. Welch, J. Med. Chem., 1992, 35, 1212 CrossRef CAS.
- B. Iselin and R. Schwyzer, Helv. Chim. Acta, 1960, 43, 1760 CrossRef.
- M. Hudlický, Reductions in Organic Chemistry, ACS Monograph 188, Washington DC, 2nd edn., 1996, pp. 35 (reduction with Al) and p. 126 (cleavage of sulfonamides) Search PubMed.
- F. Weygand and E. Frauendorfer, Chem. Ber., 1970, 103, 2437 Search PubMed.
- J. O. Osby, M. G. Martin and B. Ganem, Tetrahedron Lett., 1984, 25, 2093 CrossRef CAS and references cited therein.
- (a) L. Grehn and U. Ragnarsson, Angew. Chem., 1985, 97, 519 ( Angew. Chem., Int. Ed. Engl. , 1985 , 24 , 510 ) CAS; (b) S. K. Davidsen, P. D. May and J. B. Summers, J. Org. Chem., 1991, 56, 5482 CrossRef CAS; (c) B. M. Kim, B. E. Evans, K. F. Gilbert, C. M. Hanifin, J. P. Vacca, S. R. Michelson, P. L. Darke, J. A. Zugay, E. A. Emini, W. Schleif, J. H. Lin, I.-W. Chen, K. Vastag, P. S. Anderson and J. R. Huff, Bioorg. Med. Chem. Lett., 1995, 5, 2707 CrossRef CAS; (d) M. D. Ennis, N. B. Ghazal, R. L. Hoffman, M. W. Smith, S. K. Schlachter, C. F. Lawson, W. B. Im, J. F. Pregenzer, K. A. Svensson, R. A. Lewis, E. D. Hall, D. M. Sutter, L. T. Harris and R. B. McCall, J. Med. Chem., 1998, 41, 2180 CrossRef CAS; (e) N. Kojima, N. Minakawa and A. Matsuda, Tetrahedron, 2000, 56, 7909 CrossRef CAS.
- (a) Y. Takeuchi, M. Kamezaki, K. Kirihara, G. Haufe, K. W. Laue and N. Shibata, Chem. Pharm. Bull., 1998, 46, 1062 Search PubMed; (b) S. Yoshifuji, K. Tanaka and Y. Nitta, Chem. Pharm. Bull., 1985, 33, 1749 Search PubMed; (c) N. Sakura, K. Hirose and T. Hashimoto, Chem. Pharm. Bull., 1985, 33, 1752 Search PubMed; (d) N. Sakura, K. Hirose and T. Hashimoto, Chem. Pharm. Bull., 1986, 34, 1506 Search PubMed.
- J. Symerský, P. Maloň, L. Grehn and U. Ragnarsson, Acta Crystallogr., Sect. C, 1990, 46, 683 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: reductive cleavage of N-substituted aromatic amides as tert-butyl acylcarbamates. See http://www.rsc.org/suppdata/p1/b1/b107330n/ |
| This journal is © The Royal Society of Chemistry 2002 |