The effect of oxygen on the regioselectivity in the rhodium catalysed hydrosilylation of 1,3-dienes†
Magnus
Gustafsson
and
Torbjörn
Frejd
*
Organic Chemistry, Centre for Chemistry and Chemical Engineering, Lund University, P.O. Box 124, S-221 00 Lund, Sweden. E-mail: torbjorn.frejd@orgk1.lu.se
First published on 6th December 2001
Abstract
The regioselectivity of the hydrosilylation of substituted 1,3-dienes catalysed by several rhodium complexes in the presence and absence of oxygen was studied. In addition to the already known accelerating effect, the presence of oxygen strongly affected the product distribution. For 2-substituted 1,3-dienes in the presence of oxygen the regioselectivity was in the range of 1 ∶ 6 to 1 ∶ 10 in favour of the head-product, while the absence of oxygen changed the ratios to 1 ∶ 1 to 3 ∶ 1 in favour of the tail-product. When HSiPh3 was used in the presence of oxygen a single isomer was isolated in 87% yield, while in the absence of oxygen a mixture of products was produced. Control experiments indicated that a heterogeneous/colloidal catalytic system may be responsible for the preferred head-product formation.
Introduction
Allylic silanes are useful synthetic intermediates in organic synthesis, for example as components in the Hosomi–Sakurai reaction and the [3 + 2] cycloaddition of unsaturated ketones to give five membered rings.1–3 The silanes may also act as masked hydroxy groups as they can be oxidized to give alcohols according to the procedure developed by Tamao et al. and Fleming et al.4,5 The transition metal-catalysed hydrosilylation of alkenes is an attractive way of attaching silicon substituents to the carbon skeleton, e.g. the hydrosilylation of 1,3-dienes may give allylic silanes via a 1,4-addition of the hydrosilane. When an unsymmetrical diene is used, four different isomers may be produced via the 1,4-addition (Scheme 1).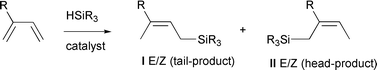 | ||
| Scheme 1 Hydrosilylation of an unsymmetrical diene. | ||
Depending on the catalyst, various E–Z mixtures of I (tail-product) and II (head-product) may be produced. It has been shown that when rhodium- or palladium-based catalysts are used, a mixture of I and II is obtained via syn-addition of the hydrosilane.6 The isomer ratio is dependent on many factors such as solvent, hydrosilane and temperature but mainly on the catalyst metal. For example, Pd-based catalysts give predominantly the (Z)-II isomer, while Rh-based catalysts can give either (E)-I or (Z)-II as the main product.7,8
Oxygen and organic peroxides have been used to enhance the catalytic activity of certain metal complexes such as Rh, Ru and Pt.9–13 Parish et al. showed that under strictly deoxygenated conditions RhCl(PPh3)3 was inactive as a catalyst in the hydrosilylation of alkenes and alkynes. When a catalytic amount of oxygen or a peroxide was present, the complex became catalytically active. This co-catalytic effect was explained as a result of oxidation of a phosphine ligand to give an unsaturated rhodium complex (Scheme 2).9
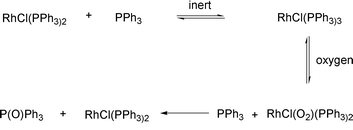 | ||
| Scheme 2 Activation of Wilkinson's catalyst by oxygen.9 | ||
A similar displacement of a coordinated ligand has been observed in the tert-butylhydroperoxide (TBHP)-initiated oxidation of [Rh(CO)Cl(PPh3)2] to give [RhCl(PPh3)2]2 and carbon dioxide.14 It is also known that several transition metals form discrete complexes with oxygen thereby changing the catalytic properties of the metal.15–18
Lewis et al. have pointed out that in many homogeneous catalytic systems containing Rh, Pt, Ir and Pd complexes the actual catalytic species is a colloid.16,19–21 Whether the catalyst is homogeneous or colloidal, electronic effects of the coordinated ligands such as O2 would be expected, which was stressed by Lewis et al. for the platinum catalysed hydrosilylation of olefins.16
Our earlier applications of catalytic 1,3-hydrosilylations resulted in only modest regiocontrol.22 Moreover, in trying to improve the selectivity we encountered considerable irregularities; the regioselectivity varied strongly from experiment to experiment for no apparent reason. During our studies, we also observed results that deviated somewhat from those in the literature. Ojima et al. had previously reported a 72 ∶ 28 ratio between I and II, in the RhCl(PPh3)3 catalysed hydrosilylation of isoprene with HSiMe2Ph.7,23 However, we observed that this reaction gave essentially only I (>99%) under certain conditions, which unfortunately could not be consistently reproduced. After extensive experimentation we found that the presence of oxygen had a considerable effect on the regiochemistry.
In this paper we wish to report the effect of molecular oxygen on the regioselectivity in the Rh-catalysed, and in one case the Ir-catalysed, hydrosilylation of unsymmetrical 1,3-dienes.
Results and Discussion
The results of the hydrosilylation of isoprene under an argon (Ar-conditions) and an oxygen atmosphere (O2-conditions), respectively, are shown in Table 1 for a number of different silanes. Under oxygen-free conditions our results were in good agreement with those of Ojima et al.7,23 The isomer ratio I ∶ II was 75 ∶ 25 for HSiMe2Ph. In these experiments the solvent was thoroughly degassed by purging with argon.| Entry | Hydrosilane | Conditions | Ratio I ∶ II | Yield (%) |
|---|---|---|---|---|
| a The reproducibility in a number of experiments was within ± 2%. b Mixture of several products, presumably via 1,2- and 1,4-additions. Several signals in the olefin region in the 1H NMR spectrum were observed. | ||||
| 1 | HSiEt3 | 80 °C, 15 h, Ar | 55 ∶ 45 | >90 |
| 2 | HSiEt3 | 80 °C, 2 h, O2 | 15 ∶ 85 | >90 |
| 3 | HSiMe2Ph | 80 °C, 2 h, Ar | 75 ∶ 25a | 92 |
| 4 | HSiMe2Ph | 80 °C, 2 h, O2 | 10 ∶ 90a | 94 |
| 5 | HSiPh3 | 110 °C, 12 h, Ar | Mixtureb | 85 |
| 6 | HSiPh3 | 110 °C, 12 h, O2 | 5 ∶ 95 | 87 |
| 7 | HSi(OMe)3 | 90 °C, 4 h, Ar | 76 ∶ 24 | 85 |
| 8 | HSi(OMe)3 | 90 °C, 4 h, O2 | 13 ∶ 87 | 84 |
| 9 | HSi(OMe)3 | 110 °C, 4 h, Ar | 55 ∶ 45 | >80 |
| 10 | HSi(OMe)3 | 110 °C, 4 h, O2 | 10 ∶ 90 | >80 |
The experiments under an oxygen atmosphere were standardised as follows: O2 was bubbled through a suspension (heptane) or a solution (benzene) of RhCl(PPh3)3 for 30 seconds. The reddish mixtures were then heated at about 90 ° C for 60 min. During this time the colour changed to yellow–orange in benzene (homogeneous) and pale yellow in heptane (heterogeneous). No further investigation were made to explain these colour changes. After cooling, the appropriate hydrosilane and diene were added. Heating this mixture gave products which were analysed by GLC. All hydrosilanes tested showed the same general trend regarding isomer distribution under the various conditions. Under an inert atmosphere isomer I was favoured (at worst a 1 ∶ 1 mixture was obtained), while II predominated under O2-conditions. The ratio ranged from 1 ∶ 6 (HSiEt3, Table 1, entry 2) to 1 ∶ 9 (HSiMe2Ph, Table 1, entry 4). When HSiPh3 (Table 1, entry 6) was used the only isolated product was II (87%).
Other substituted dienes also gave II as the major product under O2-conditions, while complex mixtures of 1,2- and 1,4-addition products were produced under Ar-conditions (Table 2). Both myrcene (Table 2, entry 1) and epoxymyrcene (Table 2, entry 2) as well as isoprene (Table 1, entry 3) gave a similar 75 ∶ 25 isomer ratio between I and II under Ar-conditions, while under O2-conditions a 1 ∶ 9 ratio was observed for all three substrates.
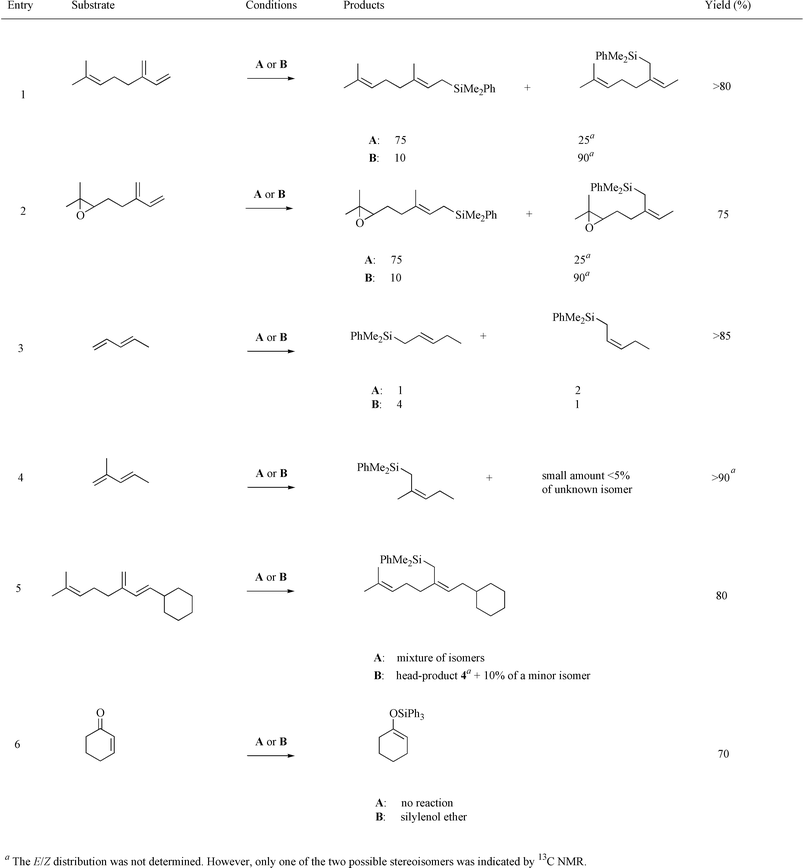
|
In all cases studied, under O2-conditions, the Z-configuration of the allylic silanes was observed except for penta-1,3-diene, which gave a considerable amount of the E-isomer and also a change in stereoisomer distribution from E–Z 1 ∶ 2 under Ar-conditions to E–Z 4 ∶ 1 under O2-conditions (Table 2, entry 3). The 1,3-disubstituted butadiene 3, synthesised from 1 in 74% overall yield via a HEW–Wittig reaction sequence (Scheme 3), gave under Ar-conditions a mixture of isomers, while the head-product 4 was formed as the major isomer in 80% yield (Table 2, entry 5) under O2-conditions. An interesting observation was that cyclohexenone was hydrosilylated to give the silyl enol ether with a sterically hindered silane such as HSiPh3 (Table 2, entry 6) in the presence of oxygen. Under Ar-conditions Wilkinson's catalyst was previously (also in our hands) shown to be ineffective in this transformation and only one successful report has, to our knowledge, appeared in the literature.24 Further substrates are under investigation.
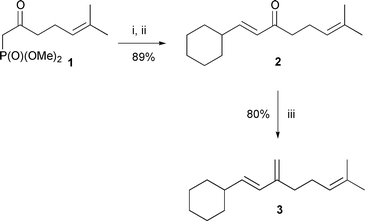 | ||
| Scheme 3 Synthesis of triene 3: i, NaH, DME, 0 °C; ii, cyclohexanecarbaldehyde, 70 °C; iii, MePPh3+Br−, BuLi, THF, 50 °C. | ||
Several other catalytic systems followed the general trend in isomeric distribution under the O2-conditions (Table 3), i.e.II was the major product. It should be noted that when CO-containing rhodium complexes such as RhCl(CO)(PPh3)2 (Table 3, entry 2) and RhH(CO)(PPh3)3 (Table 3, entry 7) or the RhIII complexes Rh(acac)3 (Table 3, entry 4) and RhCl3·H2O (Table 3, entry 5) were used, isomer II was the main product even under Ar-conditions in the hydrosilylation of isoprene with HSiMe2Ph. Under O2-conditions II predominated even more. Despite the low solubility of RhCl3·H2O in the solvents used, a good yield of the corresponding allylic silane was achieved.
| Ratio of I ∶ II | ||||
|---|---|---|---|---|
| Entry | Catalyst | Inert | Oxygen | Yield I + II (%) |
| 1 | RhCl(PPh3)3 | 75 ∶ 25 | 10 ∶ 90 | >99 |
| 2 | RhCl(CO)(PPh3)2 | 33 ∶ 67 | 14 ∶ 86 | >90 |
| 3 | RhCl(CS)(PPh3)2 | 70 ∶ 30 | 13 ∶ 87 | >90 |
| 4 | Rh(acac)3 | >5 ∶ 95 | >5 ∶ 95 | >90 |
| 5 | RhCl3·H2O | 28 ∶ 72 | 14 ∶ 86 | >90 |
| 6 | RhH(PPh3)4 | 75 ∶ 25 | 11 ∶ 89 | >90 |
| 7 | RhH(CO)(PPh3)3 | 10 ∶ 90 | 10 ∶ 90 | >90 |
| 8 | Ir(COD)2Cl2 | 50 ∶ 50 | 20 ∶ 80 | >90 |
The metalhydrido complex RhH(PPh3)4 (Table 3, entry 6) behaved like Wilkinson's catalyst and gave a ratio of 75 ∶ 25 between I and II under Ar-conditions. Under O2-conditions the ratio changed to 10 ∶ 90.
An interesting observation was that, under Ar-conditions, the thiocarbonyl complex RhCl(CS)(PPh3)2 (Table 3, entry 3) gave isomer I as the main product, while the carbonyl analogue gave II. Under O2-conditions the isomer ratios were essentially the same for both of these catalysts. The iridium complex [Ir(COD)Cl]2 was less selective, but also with this catalyst we observed a predominance of II under O2-conditions (Table 3, entry 8).
At this point a proper mechanistic study has not been undertaken, although a few remarks can be made: it is known that RhCl(PPh3)3 in methylene chloride reacts with oxygen to give two complexes, [RhCl(O2)(PPh3)3·2CH2Cl2] and [RhCl(O2)(PPh3)2·CH2Cl2]2.17,18 These complexes gave in our hands isomer I as the major product in the reaction between isoprene and dimethylphenylsilane under an inert atmosphere. This excluded that these complexes were the major catalytic species under O2-conditions. The change in the Rh ∶ PPh3 ratio due to oxidation of PPh3 to P(O)Ph3 was probably not responsible for the selectivity either, as shown by the following results. Addition of PPh3 to [RhClCOD]2 in lower ligand to metal ratios than 3 ∶ 1 still gave I as the major isomer; addition of 0, 2, 4 or 6 equivalents of the phosphine with respect to the Rh-complex gave I and II in the ratios 50 ∶ 50, 58 ∶ 42, 74 ∶ 26 and 75 ∶ 25, respectively. The lower concentration of PPh3 could explain a part of the observed selectivity change, but in the light of the fact that even the phosphine-free Ir(COD)2Cl2 gave more of isomer II under the O2-conditions, this is probably not the sole reason.
The question of radical reactions was also considered. However, the presence of a radical scavenger (galvinoxyl) gave no inhibition, which should exclude a radical chain mechanism. Still, a metal centered radical mechanism cannot be excluded, but it is not likely to operate, since the use of (Me3Si)3SiH, introduced by Giese et al. as an efficient radical generating species and also used in radical hydrosilylation of alkenes,25 did not lead to any consumption of the silane in the hydrosilylation of isoprene under O2-conditions.
As a further mechanistic possibility colloidal catalysis was considered and examined as follows. The mercury inhibition test for colloidal catalysis26,27 resulted in a moderate change of the product ratio between I and II in the hydrosilylation of isoprene with HSiMe2Ph from 17 ∶ 83 in the absence of mercury to 38 ∶ 62 in its presence. A sluggish conversion of the starting materials in the reaction which contained mercury, indicated that a more active heterogeneous/colloidal system was inhibited by the mercury. As total conversion of the starting materials was achieved, a parallel homogeneous system is likely to be operating.
Next, a rhodium colloid (“rhodium red” 2.5 nm amorphous particles) was prepared according to a procedure published by Lewis et al.20 By using this colloid as a catalyst in the hydrosilylation of isoprene, II was formed as the major isomer, again indicating that the active catalyst could be either a colloid or a heterogenous phase. However, Crivello and Fan showed that substrates containing both an alkene and an epoxide underwent ring-opening polymerization in the presence of a naked collolidal rhodium catalyst,28 which indicates that in our experiments such a colloid should not be acting since we obtained a good yield of the hydrosilylation product of epoxymyrcene (Table 2, entry 2).
The suggestion by Berberova et al.29,30 that transition metal catalysed hydrosilylation (platinum) may proceed via a radical cation is probably not true in our case. Catalysts of the platinum group rapidly oxidise hydrosilanes to their corresponding radical cations. In the absence of a substrate, fast evolution of hydrogen gas usually occurs and the hydrosilane is consumed. However, in our case, addition of the hydrosilane before the substrate did produce the allylic silane in high yield. Moreover, no evolution of hydrogen gas was observed. Further, the insignificant inhibition of the reaction using a radical scavenger suggests that the main reaction does not involve radicals.
Conclusions
We observed that the rhodium catalysed hydrosilylation of 2-substituted-1,3-dienes under an oxygen atmosphere gave the head-product as the major isomer, while the oxygen free conditions gave mainly the tail-product. To our knowledge this is the first observation that the presence of oxygen can change the product distribution in hydrosilylations, although its activating effect was known. In addition, the Rh/oxygen procedure may be an efficient way of synthesising sterically hindered allylsilanes and silyl enol ethers and be a useful catalytic system in polymer chemistry for the hydrosilylation of unsaturated polymers.Although no rigorous investigation has been made regarding the nature of the catalytic species, it seems likely that the head-selective catalyst is of colloidal/heterogeneous nature, resembling the effects of soluble RhIII-complexes.
Application of the Rh/oxygen conditions in the hydrosilylation of more highly functionalised structures will be reported elsewhere.
Experimental
All reactions were carried out under either an oxygen or an argon atmosphere. The hydrosilanes and [RhCl3·3H2O] were used as delivered without further purifications. [Rh2Cl2(COD)2],31 [Rh2Cl2(PPh3)4],32 [Rh(PPh3)3Cl],32 [RhCl(CO)(PPh3)2],33 [RhCl(CS)(PPh3)2],34 [RhH(PPh3)4],35 [RhH(CO)(PPh3)3],35 (Rh(acac)3)36 and (Ir2Cl2(COD)2)37 and epoxymyrcene38 were prepared according to literature procedures. All other dienes were commercially available. 1H NMR and 13C NMR spectra were recorded on a Varian XL 300 or on a Bruker DRX 400 NMR spectrometer. Coupling constants J are given in Hz. IR analyses were performed on a Perkin-Elmer 254 spectrophotometer. GLC analyses were performed on a Varian 3400 gas chromatograph using a SPB-5 column (Supelco, 30 m × 0.25 mm id × 0.25 μm thickness). The E–Z distribution of the products in Table 3 were not determined. However, only one of the two possible stereoisomers was indicated by 13C NMR.The reactions were carried out using the same general methods as detailed below for the hydrosilylation of isoprene with HSiMe2Ph (Method A: Ar-conditions, Method B: O2-conditions). The reaction vessels used in the catalytic runs were thoroughly washed with acid and base to minimize the risk of contamination of the finely divided rhodium particles. A blank reaction (using the standard conditions, see below but without added catalyst) confirmed that no reaction took place in the absence of added catalyst.
Typical conditions for the hydrosilylations
(Z)-1-(Triphenylsilyl)-2-methylbut-2-ene
(Table 1, entry 6, method B). The (Z)-configuration was determined by NOE spectroscopy showing the contact between H-1 and H-4. δH 1.25 (3 H, d, J 6.7), 1.57 (3 H, s), 2.42 (2 H, s), 5.15 (1 H, q, J 6.7), 7.46–7.35 (9 H, m) and 7.61–7.55 (6 H, m); δC 13.7, 19.9, 26.5, 118.4, 127.8, 129.5, 135.2, 135.9; mp 49–51 °C. HRMS (CI mode, CH4): C23H24Si (M); found: m/z 328.1647. Calc.: m/z 328.1647.The following substances were prepared according to the general procedures described above and isolated as mixtures of regio isomers. The ratio between I and II was determined by GLC (Tables 1 and 2): 1-[dimethyl(phenyl)silyl]-3-methylbut-2-ene,7 1-triethylsilyl-3-methylbut-2-ene,39 1-trimethoxysilyl-3-methylbut-2-ene, 1-triethoxysilyl-3-methylbut-2-ene,7 (Z)-1-[dimethyl(phenyl)silyl]-2-methylbut-2-ene,7 (Z)-1-triethylsilyl-2-methylbut-2-ene,39 (Z)-1-trimethoxysilyl-2-methylbut-2-ene, (Z)-1-triethoxysilyl-2-methylbut-2-ene,40,41 (E)-3,7-(dimethyl)-1-[dimethyl(phenyl)silyl]octa-2,6-diene,7 (Z)-3-[dimethyl(phenyl)silylmethyl]-7-methylocta-2,6-diene,7 1-[dimethyl(phenyl)silyl]pent-2-ene (mixture of stereoisomers),42 (Z)-1-[dimethyl(phenyl)silyl]-2-methylpent-2-ene and 1-(triphenylsilyloxy)cyclohexene.24
(Z)-2,3-Epoxy-6-[dimethyl(phenyl)silylmethyl]-2-methyloct-6-ene
(Table 2, entry 2, method B). δH 0.32 (6 H, s), 1.20 (3H, s), 1.27 (3 H, s), 1.46 (3 H, d, J 7.3), 1.50–1.67 (2 H, m), 1.74 (2 H, d, J 6.0), 1.87–2.05 (2 H, m), 2.60 (1 H, t, J 7.2), 5.15 (1 H, q, J 7.3), 7.33–7.37 (3 H, m) and 7.50–7.55 (2 H, m); δC −2.4, −2.3, 13.9, 18.7, 20.4, 24.9, 27.5, 35.6, 58.3, 64.2, 116.5, 127.8, 129.0, 133.5, 136.3 and 139.4. HRMS (CI mode, CH4): C18H28OSi (M); found: m/z, 288.1914. Calc.: m/z 288.1909.(E)-2,3-Epoxy-8-[dimethyl(phenyl)silyl]-2,6-dimethyloct-6-ene
(Table 2, entry 2, method A). δH 0.26 (6 H, s), 1.25 (3 H, s), 1.29 (3 H, s), 1.51 (2 H, s), 1.66 (3 H, d, J 8.4), 2.05–2.20 (2 H, m), 2.68 (1 H, t, J 6.4), 5.22 (1 H, t, J 8.4), 7.33–7.38 (3 H, m) and 7.50–7.55 (2 H, m); δC −3.2, −3.2, 15.8, 17.8, 18.7, 24.9, 27.8, 36.6, 58.2, 64.1, 120.2, 127.7, 128.9, 132.3, 133.5 and 139.1.Dimethyl 6-methyl-2-oxohept-5-enylphosphonate 143
A solution of n-BuLi (22 mL, 35 mmol, c = 1.6 M) in hexane was slowly added to a stirred solution of dimethyl methylphosphonate (4.4 g, 35 mmmol) in dry THF (50 mL) under an atmosphere of argon at −65 °C. After 15 min, a solution of ethyl 5-methylhex-4-enoate (3.0 g, 19 mol) in dry THF (10 mL) was added and the reaction mixture was stirred at this temperature for another 60 min. Ether (100 mL) and brine (50 mL) were then added and after acidification with 2 M HCl the phases were separated. The aqueous phase was extracted with ether (2 × 50 mL) and the combined organic phases were washed with brine (2 × 50 mL) and dried (Na2SO4). Filtration and concentration of the solvent at reduced pressure afforded crude 1. Column chromatography (EtOAc) gave 1 (2.7 g, 62%) as a colourless oil; Rf 0.23 (EtOAc); δH 1.59 (3 H, br s), 1.65 (3H, d, J 1.1), 2.24 (2 H, m), 2.62 (2 H, t, J 7.3), 3.06 (2 H, d, J 22.7), 3.75 (3 H, s), 3.78 (3 H, s) and 5.0 (1 H, apparent tq, J 7.2 and 1.4); δC 17.8, 22.3, 25.8, 40.9, 42.1, 44.3, 53.2 (d, J 7.0), 122.4, 133.2, 201.8 (d, J 6.0). HRMS (CI mode, CH4): C10H19O4P (M); found: m/z, 234.1020. Calc.: m/z 234.1021. (Analytical data were missing in ref. 43.)(1E)-1-Cyclohexyl-7-methyl-3-oxoocta-1,6-diene 2
NaH (0.34 g, 8.5 mmol, 60% in mineral oil) was added to a solution of 1 (2.0 g, 8.5 mmol) in dry DME (80 mL) under argon at 0 °C. The cooling bath was removed, and the mixture stirred for 10 min, after which the temperature was raised to 70 °C. A solution of cyclohexanecarbaldehyde (0.96 g, 8.5 mmol) in dry DME (5 mL) was then added rapidly. The reaction mixture was stirred at 70 °C for 10 min and then cooled. Ether (80 mL) was added and the solution was washed with brine (2 × 50 mL) and dried (Na2SO4). Filtration and evaporation of the solvent followed by column chromatography (heptane–EtOAc 5 ∶ 1) of the residue afforded 2 (1.7 g, 89%) as a colourless oil. Rf 0.27 (heptane–EtOAc 5 ∶ 1); δH 1.37–1.08 (5 H, m), 1.62 (3 H, s), 1.68 (3 H, s), 1.81–1.68 (5 H, m), 2.14 (1 H, m), 2.29 (2 H, m), 2.56 (2 H, m), 5.10 (1 H, apparent tq, J 7.2 and 1.4), 6.04 (1 H, dd, J 16.0 and 1.4) and 6.76 (1 H, dd, J 16.0 and 6.8); δC 17.9, 23.2, 25.9, 25.9, 26.1, 32.0, 40.3, 40.8, 123.2, 128.0, 132.8, 152.5 and 201.1. HRMS (EI+): C15H24O (M); found: m/z 220.1826. Calc.: m/z 220.1827.(1E)-1-Cyclohexyl-7-methyl-3-metyleneocta-1,6-diene 3
n-Butyllithium (3.8 mL, 6.1 mmol, 1.6 M in hexane) was added to a slurry of methyltriphenylphosphonium bromide (2.2 g, 6.1 mmol) in dry THF (30 mL) under argon. The reaction mixture was stirred at room temperature for 10 min. Then the temperature was raised to 50 °C and 2 (1.3 g, 5.8 mmol) in dry THF (10 mL) was added rapidly. The mixture was stirred at 50 °C for 10 min and then at 20 °C for 30 min. Ether (50 mL) and brine (30 mL) were added followed by separation of the phases. The organic phase was washed with brine (2 × 30 mL), dried (Na2SO4), filtered and concentrated at reduced pressure. Column chromatography of the residue (heptane–EtOAc 9 ∶ 1) afforded 3 as a colourless oil (1.1 g, 83%). Rf 0.55 (heptane–EtOAc 9 ∶ 1); δH 1.35–1.05 (5 H, m), 1.62 (3 H, s), 1.71 (3 H, s), 1.78–1.60 (5 H, m), 2.0 (1 H, m), 2.25–2.13 (4 H, m), 4.87 (1 H, br s), 4.91 (1 H, d, J 2.0), 5.17 (1 H, m), 5.67 (1 H, dd, J 16.0 and 7.0), 6.04 (1 H, d, J 16.0); δC 17.9, 25.9, 26.3, 27.2, 32.5, 33.2, 41.2, 113.3, 124.6, 129.6, 131.8, 136.2, 146.5. HRMS (EI+): C16H26 (M); found: m/z 218.2031. Calc.: m/z 218.2035.(2Z)-1-Cyclohexyl-7-methyl-3-[dimethyl(phenyl)silylmethyl]octa-2,6-diene 4
A catalyst solution was prepared according to the general description method B and then 3 (100 mg, 0.46 mmol) and HSiMe2Ph (63 mg, 0.46 mmol) were added sequentially. The reaction mixture was heated at 90 °C for 30 min. After cooling, ether (20 mL) and brine were added, the phases were separated and the organic phase was washed with brine (2 × 10 mL) and dried (Na2SO4). Evaporation of the solvent under reduced pressure and filtration through a short plug of silica afforded a mixture of two products 4 and a minor isomer (9 ∶ 1 ratio) in 80% yield. Rf 0.77 (heptane).Major isomer 4: δH 0.32 (6 H, s), 0.87 (2 H, m), 1.30–1.11 (4 H, m), 1.59 (3 H, br s), 1.69 (3 H, d, J 1.1), 1.76 (2 H, br s), 1.78–1.62 (7 H, m), 1.89 (2 H, apparent t, J 7.2), 2.06 (2 H, apparent q, J 7.3), 5.07 (2 H, m), 7.39–7.34 (3 H, m), 7.65–7.50 (2 H, m); δC −2.0, 17.9, 20.7, 26.0, 26.7, 26.9, 27.1, 33.5, 36.5, 38.9, 39.4, 121.6, 124.8, 127.9, 129.1, 131.4, 133.7, 136.4 and 139.9. HRMS (EI+): C24H38Si (M); found: m/z 354.2735. Calc.: m/z 354.2743.
Minor isomer: δC −1.4.1, 23.1, 26.0, 26.4, 26.5, 33.2, 33.4, 33.6, 38.9, 39.7, 40.6,40.8, 125.1, 127.8, 128.8, 129.0, 131.2, 133.8, 135.7.
The dependence of the regioselectivity on the PPh3 ∶ Rh ratio
Different quantities (n) of PPh3 (n = 0, 0.04, 0.08, and 0.12 mmol) were added to a solution of [RhClCOD]2 (10 mg, 0.02 mmol) in benzene (5 mL). The vessels were supplied with H2 (3 atm) for 1 h and were then degassed with argon. Next, HSiMe2Ph (0.20 g, 1.5 mmol) and isoprene (0.21 mL, 2.2 mmol) were added and these reaction mixtures were heated at 60 °C for 1 h and then worked up as described above. The chromatographed products were all obtained in quantitative yields. The isomer ratios between I and II were as follows: for n = 0, 50 ∶ 50, n = 0.04, 58 ∶ 42, n = 0.08, 74 ∶ 26, n = 0.12, 75 ∶ 25 as analyzed by GLC.Mercury inhibition studies as a test for colloids26,27
A standard solution was prepared as follows. RhCl(PPh3) (10 mg, 0.01 mmol) in benzene (5 mL) was treated with oxygen for 30 s and then heated at 70 °C for 60 min. Part of this solution (2.5 mL) was transferred to another vessel, Hg (3.0 g) was added and the reaction was stirred for 10 min. This treatment was to remove colloids. Then isoprene (0.44 g, 6.5 mmol) and HSiMe2Ph (0.58 g, 4.2 mmol) were added to each catalyst preparation and the reaction vessels were heated at 60 °C for 2 h. The product distribution was monitored by GLC. Addition of mercury changed the isomer ratio I ∶ II from 17 ∶ 83 to 38 ∶ 62, which indicated that a heterogeneous/colloidal catalyst could be operating, at least to some extent. Also the conversion of the substrates was slower under the Hg-conditions indicating the presence of a more sluggish catalytic homogeneous system operating in parallel with a heterogeneous/colloidal system.Acknowledgements
We thank the Swedish Natural Science Research Council, the Crafoord Foundation, the Knut and Alice Wallenberg Foundation, and the Royal Physiographic Society in Lund for financial support.References
- H. Sakurai and A. Hosomi, J. Am. Chem. Soc., 1977, 99, 1673 CrossRef.
- H.-J. Knölker, P. G. Jones and J.-B. P. Pannek, Synlett, 1990, 429 CrossRef.
- Z.-H. Peng and K. A. Woerpel, Org. Lett., 2000, 2, 1379 CrossRef CAS.
- K. Tamao, T. Kakui, M. Akita, T. Iwahara, R. Kanatani, J. Yoshida and M. Kumada, Tetrahedron, 1983, 39, 983 CrossRef CAS.
- I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29 RSC.
- B. Marciniec, Comprehensive Handbook on Hydrosilylation, Pergamon Press, Oxford, 1992 Search PubMed.
- I. Ojima and M. Kumagai, J. Organomet. Chem., 1978, 157, 359 CrossRef CAS.
- A. J. Cornish, M. F. Lappert, J. J. Macquitty and R. K. Maskell, J. Organomet. Chem., 1979, 177, 153 CrossRef CAS.
- H. M. Dickers, R. N. Haszeldine, L. S. Malkin, A. P. Mather and R. V. Parish, J. Chem. Soc., Dalton Trans., 1980, 308 RSC.
- R. A. Faltynek, Inorg. Chem., 1981, 20, 1357 CrossRef CAS.
- A. D. Calhoun, K. R. Lung, T. A. Nile, L. L. Stokes and S. C. Smith, Transition Met. Chem., 1983, 8, 365 Search PubMed.
- B. Marciniec and J. Gulinski, J. Mol. Catal., 1981, 123.
- A. Onopchenko and E. T. Sabourin, J. Org. Chem., 1987, 52, 4118 CrossRef CAS.
- I. J. Harvie and F. J. McQuillen, J. Chem. Soc., Chem. Commun., 1976, 369 RSC.
- L. N. Lewis and N. Lewis, J. Am. Chem. Soc., 1986, 108, 7228 CrossRef CAS.
- L. N. Lewis, J. Am. Chem. Soc., 1990, 112, 5998 CrossRef CAS.
- M. J. Bennett and P. B. Donaldson, Inorg. Chem., 1977, 16, 1581 CrossRef CAS.
- M. J. Bennett and P. B. Donaldson, Inorg. Chem., 1977, 16, 1585 CrossRef CAS.
- L. N. Lewis and R. J. Uriarte, Organometallics, 1990, 621 CrossRef CAS.
- L. N. Lewis, R. J. Uriarte and N. Lewis, J. Mol. Catal., 1991, 66, 105 CrossRef CAS.
- L. N. Lewis, Chem. Rev., 1993, 93, 2693 CrossRef CAS.
- L. Pettersson and T. Frejd, J. Chem. Soc., Chem. Commun., 1993, 1823 RSC.
- I. Ojima and M. Kumagai, J. Organomet. Chem., 1977, 134, C6 CrossRef CAS.
- C. R. Johnson and R. K. Raheja, J. Org. Chem., 1994, 59, 2287 CrossRef CAS.
- K. J. Kulicke and B. Giese, Synlett, 1990, 91 CrossRef CAS.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 855 CrossRef CAS.
- G. M. Whitesides, M. Hackett, R. L. Brainard, J.-P. P. M. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staudt, Organometallics, 1985, 1819 CrossRef CAS.
- J. V. Crivello and M. Fan, J. Polym. Sci., Polym. Chem., 1992, 30, 1 Search PubMed.
- A. A. Khapicheva, N. T. Berberova, E. S. Klimov and O. Y. Okhlobystin, Zh. Obshch. Khim., 1985, 55, 1533 Search PubMed.
- O. Y. Okhlobystin and N. T. Berberova, Dokl. Akad. Nauk. SSSR, 1993, 332, 599 Search PubMed.
- G. Giordano and R. H. Crabtree, Inorg. Synth., 1979, 19, 88.
- J. A. Osborn and G. Wilkinson, Inorg. Synth., 1979, 19, 77.
- D. Evans, J. A. Osborn and G. Wilkinson, Inorg. Synth., 1979, 19, 79.
- M. Kubota and C. O. M. Ho, Inorg. Synth., 1990, 28, 204.
- N. Ahmad, J. J. Levison, S. D. Robinson and M. F. Uttley, Inorg. Synth., 1979, 19, 81.
- H. Awano, H. Watarai and N. Suzuki, J. Inorg. Nucl. Chem., 1979, 41, 124 Search PubMed.
- A. v. d. Ent and A. L. Onderdelinden, Inorg. Synth., 1979, 19, 91.
- K. Mori, Agric. Biol. Chem., 1974, 38, 2045 Search PubMed.
- W. Abdelgader, D. Chmielewski, F.-W. Grevels, S. Özkar and N. B. Peynircioglu, Organometallics, 1996, 15, 604 CrossRef.
- M. Brockmann, H. T. Dieck and I. Kleinwaechter, J. Organomet. Chem., 1986, 309, 345 CrossRef CAS.
- Z. M. Michalska, B. Ostaszewski and K. Strzelec, J. Organomet. Chem., 1995, 496, 19 CrossRef CAS.
- K. Fugami, J.-i. Hibino, S. Nakatsukasa, S. Matsubara, K. Oshima, K. Utimoto and H. Nozaki, Tetrahedron, 1988, 44, 4277 CrossRef CAS.
- P. Callant, L. D'Haenens and M. Vandewalle, Synth. Commun., 1984, 14, 155 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra. See http://www.rsc.org/suppdata/p1/b1/b106143g/ |
| This journal is © The Royal Society of Chemistry 2002 |