Controlled stepwise conversion of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine into 2,4,6,8-tetrasubstituted pyrimido[5,4-d]pyrimidines
Julian S.
Northen
a,
F. Thomas
Boyle
b,
William
Clegg
c,
Nicola J.
Curtin
d,
Andrew J.
Edwards
c,
Roger J.
Griffin
a and
Bernard T.
Golding
*a
aDepartment of Chemistry, Bedson Building, University of Newcastle upon Tyne, Newcastle upon Tyne, UK NE1 7RU
bCancer Therapeutic Research, ZENECA Pharmaceuticals, Mereside, Alderley Park, Macclesfield, Cheshire, UK SK10 4TG
cChemical Crystallography Unit, Department of Chemistry, Bedson Building, University of Newcastle upon Tyne, Newcastle upon Tyne, UK NE1 7RU
dCancer Research Unit, Medical School, University of Newcastle upon Tyne, UK NE1 7RU
First published on 3rd December 2001
Abstract
For the rational synthesis of 2,4,6,8-tetrasubstituted pyrimido[5,4-d]pyrimidines, required as purine mimetics, sequential nucleophilic substitutions of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine have been investigated. Reaction conditions have been devised leading to 2,4,6,8-tetrasubstituted pyrimido[5,4-d]pyrimidines with patterns of substitution denoted as abab (reaction with nucleophile 1 at C-4 and C-8, followed by nucleophile 2 at C-2 and C-6) or abac (reaction with nucleophile 1 at C-4 and C-8, nucleophile 2 at C-2 and nucleophile 3 at C-6) or abcd (reaction with nucleophile 1 at C-4, nucleophile 2 at C-8, nucleophile 3 at C-2 and nucleophile 4 at C-6). The use of low temperature, relatively dilute solution and careful addition of the amine nucleophile can control the critical first step. The third step in the production of the abcd pattern leads to two regioisomers, which have been structurally characterised by 1H NMR and a crystal structure analysis. Selected 2,4,6,8-tetrasubstituted pyrimido[5,4-d]pyrimidines were tested as inhibitors of the cyclin-dependent kinase 1 complex (cyclin B/CDK1), but none of the compounds showed significant activity.
Introduction
Pyrimido[5,4-d]pyrimidines are a relatively little studied heterocyclic system, which are of interest in the context of drug development because of their structural similarity to purines. We are especially interested in inhibitors of cyclin-dependent kinases (CDKs) and have developed purine derivatives (e.g. 2-amino-6-cyclohexylmethoxypurine 1) that inhibit the cyclin B1/CDK1 and cyclin A3/CDK2 complexes at concentrations in the low micromolar range.1 These purines are analogues of the ‘benchmark’ inhibitors olomoucine 2a and roscovitine 2b,2,3 although they bind to CDK2 in a different orientation.1 The objective of the present study was to obtain pyrimido[5,4-d]pyrimidine analogues of the purine CDK inhibitors. The synthetic access to such molecules requires that the pyrimido[5,4-d]pyrimidine scaffold be manipulated in a rational way. To this end we have investigated how to control the substitution chemistry of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine (3, TCPP).4 This compound is readily prepared from 1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8-(3H,7H)-tetraone 4.4 Besides enabling the synthesis of individual purine mimetics, a better knowledge of the chemistry of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidines would provide an opportunity for parallel or combinatorial syntheses of a diverse array of novel compounds.TCPP is the classical precursor of 2,4,6,8-tetrasubstituted pyrimido[5,4-d]pyrimidines, including the useful coronary vasodilator dipyridamole (5, Persantin).5 The substitution pattern in dipyridamole can be described as abab where a corresponds to a substituent at the 4- and 8-positions of the pyrimido[5,4-d]pyrimidine scaffold, whereas b corresponds to a substituent at the 2- and 6-positions (Fig. 1). The formation of pyrimido[5,4-d]pyrimidines with an abab pattern of substitution is the typical outcome of two stepwise substitution reactions of TCPP, employing an excess of nucleophile in each step.6,7 This is because the rates of substitution greatly favour the C-4 and C-8 positions over C-2 and C-6. For example, treatment of TCPP with an excess of benzylamine at ambient temperature
led to the formation of 4,8-bis(benzylamino)-2,6-dichloropyrimido[5,4-d]pyrimidine 6a. Further reaction of this compound with an excess of ethanolamine (2-aminoethanol) at 120 °C gave 4,8-bis(benzylamino)-2,6-bis(2-hydroxyethylamino)pyrimido[5,4-d]pyrimidine 6b (Scheme 1). The observed regioselectivity relates to the ability to delocalise the negative charge arising from attack of a nucleophile at C-2/C-6 or C-4/C-8. Attack at C-4/C-8 gives a species in which negative charge on nitrogen (e.g. N-3) can be delocalised into an adjacent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, whereas attack at C-2/C-6 does not permit this stabilising interaction.
N bond, whereas attack at C-2/C-6 does not permit this stabilising interaction.
![Conversion of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine 3 (TCPP) into 6a–6d. Reagents and conditions: (i) 4 equiv. benzylamine, THF, rt, 20 min; (ii) ethanolamine, 120 °C 12 h; (iii) 64 equiv. ethanolamine, THF, 75 °C, 6 h; (iv) 3.9 equiv. KOH, H2, 30–40 psi, 10% Pd/C, THF, 20 h.](/image/article/2002/P1/b102224p/b102224p-s1.gif) | ||
| Scheme 1 Conversion of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine 3 (TCPP) into 6a–6d. Reagents and conditions: (i) 4 equiv. benzylamine, THF, rt, 20 min; (ii) ethanolamine, 120 °C 12 h; (iii) 64 equiv. ethanolamine, THF, 75 °C, 6 h; (iv) 3.9 equiv. KOH, H2, 30–40 psi, 10% Pd/C, THF, 20 h. | ||
![Substitution patterns for pyrimido[5,4-d]pyrimidines.](/image/article/2002/P1/b102224p/b102224p-f1.gif) | ||
| Fig. 1 Substitution patterns for pyrimido[5,4-d]pyrimidines. | ||
The reactions of 4,8-bis(benzylamino)-2,6-dichloropyrimido[5,4-d]pyrimidine with nucleophiles can be carried out in a stepwise fashion to afford 4,8-bis(benzylamino)-2,6-disubstituted pyrimido[5,4-d]pyrimidines, with different substituents at C-2 and C-6, i.e. an abac substitution pattern (cf. Fig. 1). Furthermore, we have found that the reaction of TCPP with benzylamine can be controlled to give the mono-substituted product, 4-benzylamino-2,6,8-trichloropyrimido[5,4-d]pyrimidine, in acceptable yield. This compound served as a valuable starting material for the preparation of 2,4,6,8-tetrasubstituted pyrimido[5,4-d]pyrimidines with an abcd pattern of substitution, via a sequence of controlled nucleophilic substitutions of the remaining three chloro functions. The structures of the abac and abcd tetrasubstituted pyrimido[5,4-d]pyrimidines described herein were validated by spectroscopic characterisation of all compounds, and for the abcd type by an X-ray analysis of a representative end-product. The abcd pattern of substitution was of particular interest in the context of analogues of purine CDK inhibitors and so selected compounds were evaluated as inhibitors of the cyclin B/CDK1 complex.
Results and discussion
Symmetrical and non-symmetrical 4,8-bis(benzylamino)pyrimido[5,4-d]pyrimidines (abab and abac substitution pattern e.g. a = benzylamino, b = 2-hydroxyethylamino, c = H)
Various methods to access TCPP 3 have been described.4 We found that a satisfactory method is to heat an aqueous solution of 5-aminoorotic acid at 210 °C in the presence of ammonium chloride and potassium cyanate to give tetraone 4.4 This was isolated as its disodium salt, which was converted into 3 by treatment with a mixture of phosphorus oxychloride and phosphorus pentachloride. Reaction of 4 mol equivalents of benzylamine with TCPP in THF at ambient temperature rapidly gave 4,8-bis(benzylamino)-2,6-dichloropyrimido[5,4-d]pyrimidine 6a in good yield, without formation of the tri- and tetra-substituted benzylamino derivatives (Scheme 1). The procedure used was a modification of that reported,7 employing reduced quantities of benzylamine to improve the isolation and purification of compound 6a.For the synthesis of symmetrical (abab) and non-symmetrical (abac) 2,6-disubstituted 4,8-bis(benzylamino)pyrimido[5,4-d]pyrimidines, nucleophilic substitutions at the C-2 and C-6 positions were required. Heating 6a with neat ethanolamine at 100 °C gave 4,8-bis(benzylamino)-2,6-bis(2-hydroxyethylamino)pyrimido[5,4-d] pyrimidine 6b (Scheme 1). Substitution at the 2- (or -6) position of 6a with an amine should result in deactivation of the pyrimido[5,4-d]pyrimidine system to further substitution. By carefully controlling the reaction temperature and the concentration of amine used, it was possible to introduce an amino function selectively at C-2 (or C-6). Thus, when compound 6a was heated with ethanolamine in THF at 75 °C 4,8-bis(benzylamino)-6-chloro-2-(2-hydroxyethylamino)pyrimido[5,4-d]pyrimidine 6c was obtained in good yield.
The reductive dehalogenation of 6c was performed using Pd/C, KOH and dry THF in a pressure vessel under a hydrogen atmosphere of 30–40 psi.8 This method produced compound 6d cleanly and in good yield, with no apparent debenzylation (Scheme 1). Having demonstrated a selective method for the synthesis of the abac substituted pyrimido[5,4-d]pyrimidines with benzylamino substituents at C-4 and C-8, the methodology was applied to the generation of analogous p-methoxyanilino compounds. Reaction of 4-methoxyaniline with TCPP (2.1 ∶ 1 molar ratio) in THF at ambient temperature gave 4,8-bis(4-methoxyanilino)-2,6-dichloropyrimido[5,4-d]pyrimidine 7a in good yield (Scheme 2). Subsequent reaction with ethanolamine in THF selectively produced 4,8-bis(4-methoxyanilino)-6-chloro-2-(2-hydroxyethylamino)pyrimido[5,4-d]pyrimidine 7c, whereas the use of neat ethanolamine gave 4,8-bis(4-methoxyanilino)-2,6-bis(2-hydroxyethylamino)pyrimido[5,4-d]pyrimidine 7b. Reductive dehalogenation of 7c furnished 4,8-bis(4-methoxyanilino)-2-(2-hydroxyethylamino)pyrimido[5,4-d]pyrimidine 7d (Scheme 2).
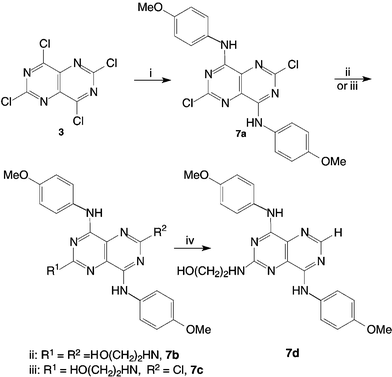 | ||
| Scheme 2 Conversion of TCPP into 7a–d. Reagents and conditions: (i) 2.1 equiv. 4-methoxyaniline, 2.4 equiv. K2CO3, THF, rt, 20 min; (ii) ethanolamine, 100 °C, 12 h; (iii) 146 equiv. ethanolamine, THF, 65 °C, 6 h; (iv) 4.6 equiv. KOH, H2, 30–40 psi, 10% Pd/C, THF, 7 days. | ||
Both 7c and 7d displayed marked fluorescence in organic solvents and were also highly photosensitive. Aerial photosensitivity has been reported for dipyridamole 5, whereby one of the piperidine rings is oxidised to give 2,6-bis(2-hydroxyethylamino)-4-(2-oxopiperidinyl)-8-piperidinylpyrimido[5,4-d]pyrimidine.9
C-4 Monobenzylaminopyrimido[5,4-d]pyrimidine analogues (abcd substitution pattern, a = benzylamino, b = e.g. methylamino, c = 2-hydroxyethylamino, d = H)
The synthesis of homopurine analogues of olomoucine or roscovitine requires selective monosubstitution of the 4 (or 8) chloro group of TCPP 3 with benzylamine. A variety of reaction parameters were investigated in order to achieve this goal. Reproducible results were obtained using a 1.1 ∶ 1 molar ratio of benzylamine to TCPP in an equal volume of THF at −78 °C, with a controlled, dropwise addition of the benzylamine solution from a syringe at a rate of 1 ml per minute. After stirring the mixture for 5 minutes, addition of water precipitated 8 (Scheme 3) in acceptable yield and sufficiently pure by TLC and HPLC analysis to be used directly.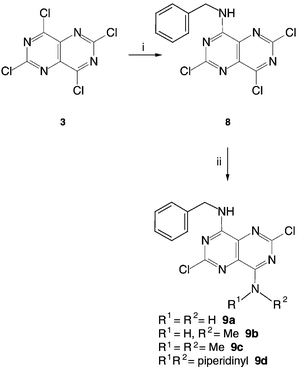 | ||
| Scheme 3 Conversion of TCPP into 8 as an intermediate in the synthesis of 9a–d. Reagents and conditions: (i) 1.14 equiv. benzylamine, K2CO3, THF, −78 °C, 15 min; (ii) excess R1R2NH , THF, 0 °C, 40 min. | ||
Treatment of the monobenzylamino derivative 8 with ammonia, methylamine, dimethylamine or piperidine in THF at 0 °C gave the corresponding 4,8-disubstituted pyrimido[5,4-d]pyrimidines 9a–d (Scheme 3). Heating each of compounds 9a–d with ethanolamine in THF afforded the corresponding trisubstituted pyrimido[5,4-d]pyrimidine derivatives 10a–d (Scheme 4). In the absence of THF and with an excess of ethanolamine the corresponding tetrasubstituted pyrimido[5,4-d]pyrimidines 11a–d were obtained.
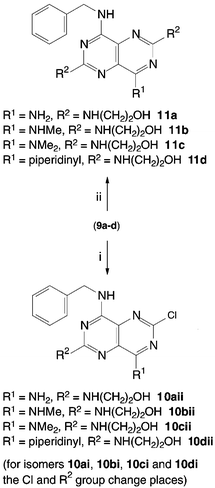 | ||
| Scheme 4 Mono and disubstitution of 9a–d with ethanolamine. Reagents and conditions: i) ethanolamine, THF, 65 °C, 6 h; ii) excess ethanolamine, 100 °C. | ||
Dehalogenation of compounds 10a–d (Scheme 5) gave the corresponding homopurines 12a–d in good yield. Only compound 11b in the 2,6-bis(2-hydroxyethylamino) series could be obtained in a pure form. Similarly, compound 12b was the only member of the dehalogenated derivatives to be obtained in a pure enough form for characterisation and biological evaluation. This was due to the instability of the compounds and difficulties encountered with purification.
![Reductive dehalogenations of compounds 10aii–dii [note these reactions were carried out on mixtures of 10ai and 10aii, 10bi and 10bii, 10ci and 10cii, 10di and 10dii leading to mixtures of 12ai and 12aii, 12bi and 12bii, 12ci and 12cii, 12di and 12dii, respectively; only the structures of the ii isomers are shown (the i isomers have the Cl or H at C-2 and the CH2CH2OH at C-6)]. Reagents and conditions: i) 6.1 equiv. KOH, H2, 10% Pd/C, THF, 20 h.](/image/article/2002/P1/b102224p/b102224p-s5.gif) | ||
| Scheme 5 Reductive dehalogenations of compounds 10aii–dii [note these reactions were carried out on mixtures of 10ai and 10aii, 10bi and 10bii, 10ci and 10cii, 10di and 10dii leading to mixtures of 12ai and 12aii, 12bi and 12bii, 12ci and 12cii, 12di and 12dii, respectively; only the structures of the ii isomers are shown (the i isomers have the Cl or H at C-2 and the CH2CH2OH at C-6)]. Reagents and conditions: i) 6.1 equiv. KOH, H2, 10% Pd/C, THF, 20 h. | ||
The reactions described for compounds 9a–d leading to 10a–d and 12a–d produced an equimolar mixture of C-2 and C-6 regioisomers, as gauged by 1H NMR analysis (Table 1). The respective isomers were separated chromatographically for compounds 10b (i and ii) and 12b (i and ii). 1H NMR analysis of the separated isomers allowed their assignment as 12bi (‘C-6 isomer’) for the less polar compound and as 12bii (‘C-2 isomer’) for the more polar compound. The assignment was based on the positions of the resonances from the N-alkyl group and benzyl methylene. Elucidation of the structures of the isomers of compound 12b (i and ii) was aided by their protonation with DCl (Table 1). The N-methyl resonance furthest upfield should correspond to the pyrimidine ring with the (2-hydroxyethylamino) group attached. In contrast, the ring bearing no substituent at C-2 or C-6 should exhibit a downfield shift for either the N-methyl or N-benzyl methylene resonance. Protonation of the pyrimido[5,4-d]pyrimidine system (most likely on an endocyclic nitrogen) resulted in a downfield shift of all resonances and especially the resonance for H-2/H-6.
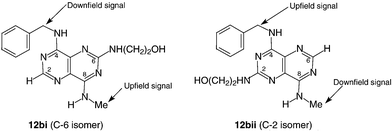
Proof of the structure of 12bii, and hence 12bi, was obtained by X-ray crystal structure analysis which confirmed compound 12bii as the C-2 isomer (for data see Fig. 2). Two conformers, anti and gauche with respect to the N(14)–C(26) (Fig. 2a) and N(7)–C(10) (Fig. 2b) bonds, were observed in the crystallographic asymmetric unit of the structure. These are linked together by a network of hydrogen bonds involving all eight potential O–H and N–H donors, with N atoms of the pyrimido[5,4-d]pyrimidine systems as acceptors, O⋯N and N⋯N distances ranging from 2.72 to 3.17 Å. There is also approximately parallel stacking of pyrimido[5,4-d]pyrimidine and phenyl rings in various directions in the crystal structure. This combination of hydrogen bonding and hydrophobic stacking interactions may help to explain the poor solubility of the pyrimido[5,4-d]pyrimidine analogues.
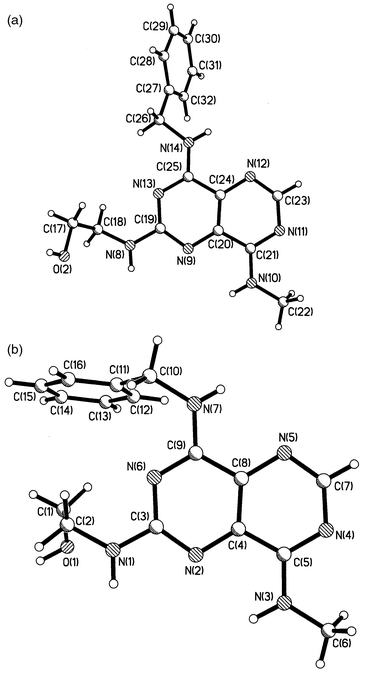 | ||
| Fig. 2 The anti (a) and gauche (b) conformers observed in the crystal structure of 12bii. | ||
All of the exocyclic C–N bonds connecting to the pyrimido[5,4-d]pyrimidine ring are relatively short [ca. 1.34 Å: N14–C25 (1.333), N10–C21 (1.333), N8–C19 (1.355); atom numbers refer to the crystal structure assignment], and are approximately the same length as the carbon–carbon double bond of ethene (1.33 Å), suggesting partial double bond character. These data are in accord with the observation of rotameric species in the 1H NMR spectra of certain compounds, in particular 8 and 9c. The spectra displayed two broad singlets for both the benzyl methylene and dimethylamino groups, each pair collapsing to a broad singlet upon heating the samples to coalescence. The corresponding figures for the C-alkyl–N bonds, by comparison, are greater [by ca. 0.1 Å: N14–C26 (1.464), N10–C22 (1.464), N8–C18 (1.450)], as would be expected for a non-conjugated carbon–nitrogen single bond.
Inhibition of cyclin B/CDK1
Three initial series of pyrimido[5,4-d]pyrimidine analogues have been synthesised in order to determine the inhibitory potential of the pyrimido[5,4-d]pyrimidine pharmacophore against cyclin B/CDK1. The activity of the cyclin B/CDK1 complex, in the presence of seven selected pyrimido[5,4-d]pyrimidines (6b–6d, 7c, 7d, 10bi, 12bi), was measured by its ability to phosphorylate a cellular substrate using radiolabelled 32P ATP.10 Difficulties were encountered in obtaining accurate data because of the low solubility of all of the compounds in aqueous media. This necessitated the inclusion of either 1% or 10% DMSO in the assay medium, although even with 10% DMSO some compounds could not be assayed at a concentration of 100 μM. It should be noted that a 10 μM concentration of the reference standard olomoucine gave 64% inhibition (IC50 9 μM; note IC50 is the concentration of drug required to reduce the activity of the enzyme by 50%) in 1% DMSO, whereas the same concentration of drug in 10% DMSO gave 31% inhibition. The results indicated that none of the pyrimido[5,4-d]pyrimidines tested exhibited significant activity (i.e. IC50 < 100 μM) against cyclin B/CDK1.Conclusions
The work described in this paper has shown how reactions of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine with nucleophiles can be controlled to give structurally characterised tetrasubstituted pyrimido[5,4-d]pyrimidines with abab, abac or abcd substitution patterns. The methodology described in this paper should facilitate the design of pyrimido[5,4-d]pyrimidine analogues, which are more soluble in aqueous media and more active as CDK inhibitors. Furthermore, the results presented indicate how combinatorial approaches could be used for the development of biologically active compounds based on the pyrimido[5,4-d]pyrimidine scaffold.Experimental
All techniques were performed as described in earlier work,11 except where indicated below. Combustion analyses were carried out on a Carlo-Erba Instrumentazione 1106 analyser or by Butterworths Laboratories Ltd, 54–56 Waldergrave Road, Teddington, Middlesex, UK TW11 8LG. All proton and carbon nuclear magnetic resonance (1H NMR and 13C NMR) spectra were obtained in deuteriated dimethyl sulfoxide (d6-DMSO) except where indicated and exchangeable protons were validated using a deuterium oxide (D2O) ‘shake’. Carbon spectra were recorded using a Bruker AMX 500 spectrometer and are broad band decoupled. The following abbreviations are used to describe peak patterns in proton spectra: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet; and combinations thereof, e.g. dd (double doublet).The general starting material 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine and all products derived from the pyrimido[5,4-d]pyrimidine nucleus were protected from direct light sources due to their potential photosensitive nature.9 Tetrahydrofuran was distilled from sodium and benzophenone, and used immediately. Benzylamine was dried over calcium hydride and distilled. Dimethylacetamide was dried over barium oxide and distilled. Distilled solvents were stored over 4 Å molecular sieves under an inert gas atmosphere.12
2,4,6,8-Tetrachloropyrimido[5,4-d]pyrimidine 34
A solution containing 5-aminoorotic acid† (1.06 g, 6.19 mmol), potassium cyanate (2.54 g, 31.36 mmol) and ammonium chloride (3.35 g, 63.2 mmol) in water (19 ml) was stirred for 0.5 h. The mixture was heated to 140 °C whilst the water was allowed to evaporate, giving a thick paste. Heating the paste at 210 °C for 0.5 h gave a beige solid which was cooled, triturated with water and filtered. After drying, the solid was added to a boiling solution of sodium hydroxide (0.1 M, 125 ml), and the yellow solution was left to cool. The product was removed by filtration to yield pale orange crystals, which were dried and used directly (0.88 g, 73%). 3,7-Disodio-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8-(3H,7H)-tetraone (6.0 g, 25 mmol) was mixed with phosphorus oxychloride (250 ml) and phosphorus pentachloride (30 g) under nitrogen and refluxed for 15 h. Phosphorus oxychloride was removed by short-path distillation to give a brown solid to which crushed ice (350 ml) was added. The slurry was filtered and the remaining brown solid was dried and extracted with chloroform using a Soxhlet apparatus. Removal of the solvent in vacuo gave the product as a pale yellow powder which was used directly (3.44 g, 51%); λmax/nm 329 (7400), 282 (2800), 258 (12300); EIMS m/z 274 (M + 6, 11%), 272 (M + 4, 49), 270 (M + 2, 100), 268 (M+, 83).4-Benzylamino-2,6,8-trichloropyrimido[5,4-d]pyrimidine 8
To TCPP (0.10 g, 0.37 mmol) and potassium carbonate (0.10 g, 0.74 mmol) in THF (6 ml), stirred under nitrogen at −78 °C, was added benzylamine (0.04 g, 0.42 mmol) in THF (6 ml) dropwise from a syringe at a rate of 1 ml per min. A cloudy yellow solution was obtained which was allowed to warm up for 10 min. Addition of an excess of water gave a fine yellow precipitate which was extracted with ethyl acetate. After drying of the organic extract over MgSO4, removal of the solvent gave a yellow glass. Trituration with petrol, filtration and drying in vacuo gave the title compound as a pale yellow solid, which was used without further purification (0.09 g, 72%); δH 4.69–4.72 (2 H, d, J 6.1 Hz, ArCH2N), 4.72–4.81 (2 H, d, J 6.4 Hz, ArCH2N), 7.31–7.50 (5 H, m, Ar-H), 9.20 (1 H, br s, ArCH2NH), 10.19 (1 H, br s, ArCH2NH); m/z 345 (M + 6, 3%), 343 (M + 4, 23), 341 (M + 2, 73), 339 (M+, 79), 308 (3), 306 (12), 304 (18), 233 (40), 91 (100). Calculated for C13H8N5Cl3: m/z 338.98. Found 338.98.Synthesis of abab substituted-pyrimido[5,4-d]pyrimidines (a = NHR, b = Cl); standard procedure A
To TCPP (2.90–7.40 mmol) in dry THF (50–60 ml) stirred under N2 at 25 °C, was added an appropriate amine (4.0 mol equiv. or 2.1 mol equiv. with 2.40 equiv. potassium carbonate). The resulting suspension was agitated for 20 min. Addition of water (80 ml) precipitated the product. The solid was filtered off, washed with water and dried. The resulting product was either used as such or recrystallised from hot ethyl acetate.Reaction of 4-benzylamino-2,6,8-trichloropyrimido[5,4-d]pyrimidine (8) with amines; standard procedure B
To a stirred solution of 4-benzylamino-2,6,8-trichloropyrimido[5,4-d]pyrimidine (0.077 M) in dry THF (13 ml) at 0 °C was added a solution of the appropriate amine (> 2.0 mol equiv.) in THF (13 ml) over a 10 min period. In all cases a precipitate formed and the suspension was left to stir for 10–30 min. Water (20 ml) was added and the THF was removed in vacuo. Purification of the product was achieved either by filtration and washing with water or by extraction with ethyl acetate. The crude material was dried and the product was either used directly or purified by silica column chromatography.Reaction of abab and abcc substituted-pyrimido[5,4-d]pyrimidines with ethanolamine (mono substitution); standard procedure C
To the appropriate 2,6-dichloro-4,8-disubstituted-pyrimido[5,4-d]pyrimidine (0.15–1.13 mmol) in THF (5–12 ml) under nitrogen was added ethanolamine (1.25–10 ml). The resultant yellow solution was heated at 65 °C for 2–6 h (unless stated otherwise), over which period a fine precipitate of ethanolamine hydrochloride formed. The suspension was cooled and water (50 ml) was added, precipitating the product and dissolving the salt. The resultant mixture was filtered, to give an off-white solid which was retained. The filtrate was extracted with ethyl acetate and the organic layer was washed with water, dried (Na2SO4), filtered and concentrated, producing a secondary yield of product. Column chromatography and/or recrystallisation from a suitable solvent afforded the desired compound.Reaction of abab and abcc substituted-pyrimido[5,4-d]pyrimidines with ethanolamine (disubstitution); standard procedure D
The appropriate pyrimido[5,4-d]pyrimidine was slurried in ethanolamine (2–5 ml) under nitrogen and heated at 80–120 °C for 1–3 days. After cooling the solution, water (20 ml) was added and an extraction was performed using ethyl acetate (100 ml). The organic solvent was dried (Na2SO4), and removed in vacuo. Purification of the crude product was achieved either by recrystallisation from hot ethyl acetate or by column chromatography on silica, to give the product as a pale yellow solid.Reductive dehalogenation of abac and abcd substituted-pyrimido[5,4-d]pyrimidines; standard procedure E
The appropriate pyrimido[5,4-d]pyrimidine (0.23–1.10 mmol) in dry THF (20–40 ml) was added under nitrogen to a pressure vessel containing 10% Pd/C (0.1–0.24 g) and potassium hydroxide (3.86–6.07 equiv.). The flask was evacuated and charged with hydrogen before pressurising to 40 psi and agitating for 1–7 days. On completion, the flask contents were filtered through Celite and eluted with ethyl acetate (100 ml). The solvent was dried (Na2SO4) and removed in vacuo to produce a crude solid. Purification was achieved by recrystallisation from hot ethyl acetate (or ethyl acetate–petrol) and/or silica column chromatography using methanol–dichloromethane as eluents, to give the desired product as a white solid.C–H), 8.52 (1 H, br s, ArCH2NH); m/z 401 (M+, 60%), 91 (100) (Found: C 65.64; H 5.59; N 23.98; C22H23N7O requires C 65.82; H 5.77; N 24.42%).
C–H), 9.20 (2 H, br s, ArCH2NH); m/z 433 (M+, 40%), 418 (10), 402 (20) (Found: C 61.12; H 5.16; N 22.37; C22H23N7O3 requires: C 60.96; H 5.35; N 22.62%).
12bi (white solid): δH 2.97 (3 H, distorted d, J 5 Hz, NHCH3), 3.5–3.6 (4 H, m, NH(CH2)2OH), 4.66 (1 H, br t, J 5 Hz, CH2OH), 4.76 (2 H, m, ArCH2NH), 6.6 (1 H, m, NHCH3), 7.3–7.4 (5 H, m, Ar-H), 7.9–8.0 (2 H, 2 × m, ArCH2NH and NH(CH2)2), 8.17 (1 H, s, NC–H) [note on addition of D2O, the resonances at 4.66 and 7.9–8.0 disappeared, whilst those at 2.97 and 4.76 collapsed to singlets] (Found: C 58.95; H 5.78; N 29.64; C16H19N7O requires: C 59.06; H 5.89; N 30.13%).
12bii (yellow solid): δH 3.03 (3 H, distorted d, J 5 Hz, NHCH3), 3.5–3.6 (4 H, m, NH(CH2)2OH), 4.66 (1 H, m, CH2OH), 4.69 (2 H, m, ArCH2NH), 6.6 (1 H, m, NHCH3), 7.3–7.4 (5 H, m, Ar-H), 7.5 (1 H, br m, NH), 8.14 (1 H, s, NC–H), 8.4 (1 H, br m, NH) [note on addition of D2O, the resonances at 4.66 and 7.9–8.0 disappeared, whilst those at 2.97 and 4.76 collapsed to singlets] (Found: C 58.95; H 5.78; N 29.64; C16H19N7O requires: C 59.06; H 5.89; N 30.13%).
X-Ray crystallography
Crystal data for 12bii: C16H19N7O, M = 325.4, orthorhombic, space group Pbcn, a = 38.754(8), b = 7.9580(16), c = 20.954(4) Å, V = 6462(2) Å3, Z = 16 (2 independent molecules in the asymmetric unit), synchrotron radiation (λ = 0.6889 Å),13T = 160 K; conventional R(F, F2 > 2σ) = 0.0558, Rw(F2, all data) = 0.1386 for 460 parameters and 6638 unique data. Programs: standard Bruker diffractometer control and integration software, and SHELXTL.14 CCDC reference number(s) 161624. See http://www.rsc.org/suppdata/p1/b1/b102224p/ for crystallographic files in .cif or other electronic format.Acknowledgements
We thank the EPSRC and AstraZeneca Pharmaceuticals for funding this research.References
- C. E. Arris, F. T. Boyle, A. H. Calvert, N. J. Curtin, J. A. Endicott, E. F. Garman, A. E. Gibson, B. T. Golding, S. Grant, R. J. Griffin, P. Jewsbury, L. N. Johnson, A. M. Lawrie, D. R. Newell, M. E. M. Noble, E. A. Sausville, R. Schultz and W. Yu, J. Med. Chem., 2000, 43, 2797 CrossRef CAS.
- W. F. De Azvedo, S. Leclerc, L. Meijer, M. Strnad and S.-H. Kim, Eur. J. Biochem., 1997, 243, 518 CrossRef CAS.
- U. Schulze-Gahmen, J. Brandson, H. D. Jones, D. O. Morgan, L. Meijer, J. Vesely and S.-H. Kim, Proteins: Struct., Funct., Genet., 1995, 22, 378 Search PubMed.
- F. G. Fischer and J. Roch, Liebigs Ann. Chem., 1951, 572, 217 Search PubMed.
- (a) H. W. Palst, Med. Klinik, 1959, 54, 257; (b) P. Luger and J. Roch, Acta Crystallogr. Sect, C: Cryst. Struct. Commun., 1983, 39, 1454 CrossRef; (c) J. L. Ambrus, I. Stadler, M. Kulaylat, A. Koreshi and S. Akhtar, J. Medicine, 1996, 27, 21 Search PubMed.
- K. Thomae GmbH, Br. Pat. 807,206, 1959; Chem. Abstr., 1959, 53, 12317 Search PubMed.
- H. C. Barlow, K. J. Bowman, N. J. Curtin, A. H. Calvert, B. T. Golding, B. Huang, P. J. Loughlin, D. R. Newell, P. G. Smith and R. J. Griffin, Bioorg. Med. Chem. Lett., 2000, 10, 585 CrossRef CAS.
- P. N. Rylander, Catalytic hydrogenation in organic synthesis, Academic Press, New York, 1979, 235 Search PubMed.
- K. Kigasawa, S. Shimizu, K. Ohkubo, S. Hayashida and K. Ohkubo, J. Pharm. Soc. Jpn., 1984, 104, 1191 Search PubMed.
- V. Rialet and L. Meijer, Anticancer Res., 1991, 11, 1581 Search PubMed.
- B. T. Golding, A. Mitchinson, W. Clegg, M. R. J. Elsegood and R. J. Griffin, J. Chem. Soc., Perkin Trans. 1, 1999, 349 RSC.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth Heinemann Press, Oxford, 4th edn., 1996 Search PubMed.
- W. Clegg, M. R. J. Elsegood, S. J. Teat, C. Redshaw and V. C. Gibson, J. Chem. Soc., Dalton Trans., 1998, 3037 RSC.
- G. M. Sheldrick, SHELXTL manual, version 5.1, Bruker AXS Inc., Madison, Wisconsin, USA, 1997 Search PubMed.
Footnote |
| † The IUPAC name for orotic acid is 2,6-dioxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid. |
| This journal is © The Royal Society of Chemistry 2002 |