The asymmetric Bischler–Napieralski reaction: preparation of 1,3,4-trisubstituted 1,2,3,4-tetrahydroisoquinolines
Marcello
Nicoletti
,
David
O'Hagan
* and
Alexandra M. Z.
Slawin
University of St. Andrews, School of Chemistry, Centre for Biomolecular Sciences, North Haugh, St. Andrews, Fife, UK KY16 9ST. E-mail: do1@st-andrews.ac.uk
First published on 6th December 2001
Abstract
The Bischler–Napieralski reaction, which is used to prepare dihydroisoquinolines from phenylethylamides, is demonstrated by the reaction of (S)-1-alkyl-1,2-diphenylethylamides with POCl3–P2O5. The reaction generated 3-alkyl-4-phenyl-1,2-dihydroisoquinolines with stereochemical selectivities of 80–91% de depending on the alkyl and the acetamide substituents. These are the first examples of the asymmetric Bischler–Napieralski reaction where cyclisation discriminates between two identical diastereotopic aryl groups. Reduction of the resultant dihydroisoquinoline products with LiAlH4 generated the corresponding 1,2,3,4-tetrahydroisoquinolines in a stereoselective manner, carrying three stereogenic centres at C(1), C(3) and C(4).
Introduction
The Bischler–Napieralski reaction has been used widely and for many years for the preparation of isoquinoline derivatives.1,2 The reaction involves the cyclodehydration of phenylethylamides 1 as illustrated in Scheme 1. Classically the reaction is carried out by the treatment of 1 with phosphorus oxychloride (POCl3) and phosphorus pentaoxide (P2O5) in refluxing toluene. Imidoyl chlorides such as 2 have been identified2 as reaction intermediates in the process and there is evidence to suggest that these reactions proceed via an intramolecular electrophilic aromatic substitution reaction on an intermediate nitrilium salt 33 The reaction is extremely versatile and can be carried out with a variety of substituents on the aromatic ring, with α and β groups on the phenylethylamide and with various amides substituents.4–7 We have recently developed8 a synthetic method for the synthesis of a series of (S)-1-alkyl-2,2-diphenylethylamines, including 5 and 6, from their corresponding amino acids (S)-alanine and (S)-valine. These compounds are prepared in enantiomerically pure forms and in either enantiomeric series depending on the amino acid enantiomer used at the outset. The availability of 5 and 6 presented an opportunity to explore the asymmetric Bischler–Napieralski reaction by their conversion to amides 7–9. These amides possess a diphenylmethyl group. Each of the phenyl groups is diastereotopic and therefore there are two stereochemical courses available to the cyclisation, generating potentially two diastereoisomeric products for each amide substrate. In this paper we present the first study of such an asymmetric Bischler–Napieralski reaction. Asymmetric reduction of the resultant dihydroisoquinoline products 10–15 with LiAlH4 was also explored to generate 1,2,3,4-tetrahydroisoquinolines 16–21 carrying three stereogenic centres.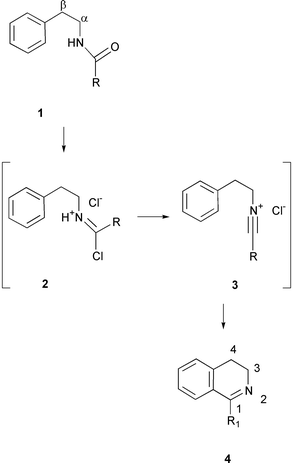 | ||
| Scheme 1 The Bischler–Napieralski reaction of 2-phenylethylamides 1 to generate the corresponding 3,4-dihydroisoquinolines 4. Salts 2 and 3 have been proposed as reaction intermediates (ref. 3). | ||
Results and discussion
The amide substrates 7–9 were prepared in a straightforward manner by reaction of either acetyl chloride or benzoyl chloride with amines 5 and 69 as shown in Scheme 2. Treatment of these substrates with POCl3–P2O5 in toluene generated the 1,3,4-trisubstituted 3,4-dihydroisoquinolines 10–15 as diastereoisomeric mixtures, but with a significant diastereoisomeric bias in each case. The yields of these reactions are poor to moderate (29–60%), a general feature of the Bischler–Napieralski reaction.1–7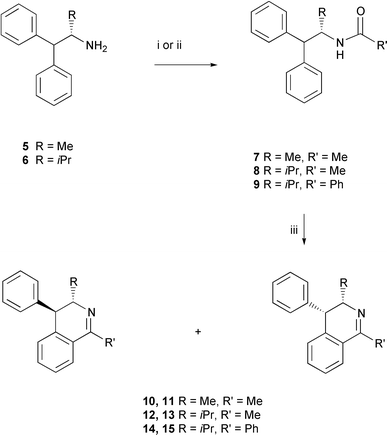 | ||
| Scheme 2 Reagents and conditions: (i) AcCl, Et3N, CH2Cl2, 12 h, 25 °C, 93–94%; (ii) benzoyl chloride, Et3N, CH2Cl2 12 h, 25 °C, 94%; (iii) POCl3, P2O5, toluene, 29–60%. | ||
For the three reactions investigated the diastereoisomeric excesses ranged from 80 to 91% de (Table 1) as determined by 1H-NMR and in the case of 12 and 13 by HPLC analyses of the product mixtures. Substrates 8 and 9, which gave products 12 (and 13) and 14 (and 15), showed the highest diastereoisomeric excesses of 86 and 91% respectively. Recrystallisation of the products gave a crystal of the predominant diastereoisomer in each case, suitable for X-ray structure analysis. The corresponding structures for 12 and 14 are shown in Figs. 1 and 2 respectively. In each case it is clear that the alkyl and aryl substituents at C(3) and C(4) have a trans relative stereochemistry. The absolute stereochemistry is secured with the knowledge that the stereogenic centre at C(3) is derived from the enantiomerically pure (S) amides 8 and 9. In the 1H-NMR spectrum of the crude reaction product 12 (and 13) derived from 8 the JH3–H4 vicinal coupling constant for the major diastereoisomer, 12, was 9.7 Hz. For the minor diastereoisomer, 13, this coupling constant was only 5.6 Hz. Similarly the major diastereoisomer 14 of the reaction products derived from amide 9 had the larger JH3–H4 vicinal coupling constant (8.7 Hz) and was assigned structure 14. The minor isomer had the smaller JH3–H4 coupling constant (5.3 Hz) consistent with structure 15. The larger coupling JH3–H4 constants for the major diastereoisomers are supported by the trans diaxial relationship between the C(3) and C(4) hydrogens as observed in the X-ray structures which were solved for 12 and 14 (Figs. 1 and 2).
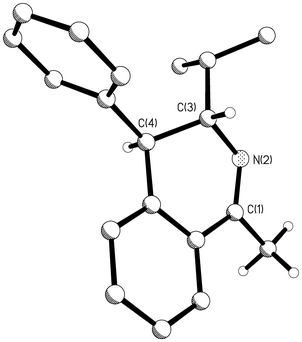 | ||
| Fig. 1 Ball and stick drawing of the molecular structure of 12 in the solid state showing the relative configuration between the substituents at C(3) and C(4) of the dihydroisoquinoline product after Bischler–Napieralski cyclisation of 8. | ||
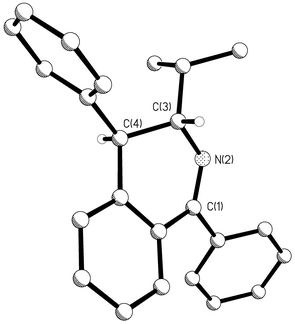 | ||
| Fig. 2 Ball and stick drawing of the X-ray generated molecular structure of 14 in the solid state showing the relative configuration between the substituents at C(3) and C(4) of the dihydroisoquinoline product after Bischler–Napieralski cyclisation of 9. | ||
| Substrate | Product | R | R′ | Yield (%) | Diastereoisomer ratios | Dea (%) | Mp/°C (major isomer) |
|---|---|---|---|---|---|---|---|
| a Diastereoisomeric excesses were calculated from the 1H-NMR analysis and in the case of 8 by chiral HPLC analysis (Chiralcel OD-H column; hexane–propan-2-ol, 99 ∶ 1; Flow 0.8 ml min−1). | |||||||
| 7 | 10, 11 | Me | Me | 29 | 9 ∶ 1 | 80 | — |
| 8 | 12, 13 | iPr | Me | 53 | 21 ∶ 1 | 91a | 65–67 (12) |
| 9 | 14, 15 | iPr | Ph | 32 | 13 ∶ 1 | 86 | 119–121 (14) |
With the 3,4-dihydroisoquinolines in hand, albeit as diastereoisomeric mixtures, LiAlH4 reduction of the imine functionality was explored to access the 1,2,3,4-tetrahydroisoquinolines and to assess the stereoselectivity of the reduction. When reduction of 12 (and 13) (21 ∶ 1) was conducted in ether or THF at reflux a similar ratio of diastereoisomeric products (20 ∶ 1) was observed to that found in the substrate from each experiment. This outcome suggested that the reduction was highly stereospecific and generated products with a trans C(1)–C(3) relative stereochemistry. The major diastereoisomer was assigned structure 16. It retained the larger JH3–H4 coupling constant of 7.1 Hz relative to that of 2.4 Hz for the minor diastereoisomeric product. A 1H-NMR difference NOE analysis was carried out and the results were again consistent with structure 16 for the major diastereoisomer. In particular, there was a strong perturbation at H(3) when the C(1) methyl hydrogen signal was irradiated, and vice versa. Also there was a strong and reciprocated NOE response between H(1) and H(4) indicating that these hydrogen atoms are on the same face of the ring system.
In the case of compound 14 (and 15) the stereoselectivity of the LiAlH4 reduction was observably solvent dependent. When the reaction was carried out in ether at reflux the products 18 (and 19) were formed in a 9 ∶ 1 ratio. Their structures were assigned again by assessing the JH3–H4 coupling constants (7.9 Hz for 18 and 3.5 Hz for 19). It is not clear why this ratio has changed from that found in the starting substrate (13 ∶ 1). It is possible that with this substrate there has been some isomerisation at C(3) prior to hydride reduction and thus isomer 20, the mirror image of 19, emerges as a minor component in the product mixture. This isomer is clearly not resolvable from 19 by 1H-NMR, and therefore the change in bias from 13 ∶ 1 to 9 ∶ 1 may reflect the generation of 20 as a minor component. The predominant products have the alkyl or aryl group at C(1) trans to the C(3) isopropyl group in the resultant tetrahydroisoquinolines.
When the reduction was carried out in THF at reflux then a new diastereoisomer was observed by 1H-NMR as a significant product (Scheme 3), in addition to 18 and 19–20. This new diastereoisomer was assigned structure 21 on the basis of a JH3–H4 coupling constant of 10.3 Hz and 1H-NMR NOE difference analysis and represents hydride delivery to the Re face of imine 14. The stereochemical outcome of these reactions in ether indicates that the LiAlH4 reduction was highly stereoselective and essentially gave exclusive delivery of hydride to the Re face of the imine substrates 12 and 14. We have no definitive hypothesis for this, however it may be assumed that lithium will co-ordinate to the imine nitrogen in an anti manner to the C(3) isopropyl group covering the lower (Si) face of the imines 10–15 (Scheme 2). Aluminium hydride will then be more easily presented to the top (Re) face of the imine. Perhaps in THF, lithium is more readily sequestered by the solvent, promoting an increased accessibility of the aluminium hydride to the Si face of 14 and thus 21 emerges as a significant product.
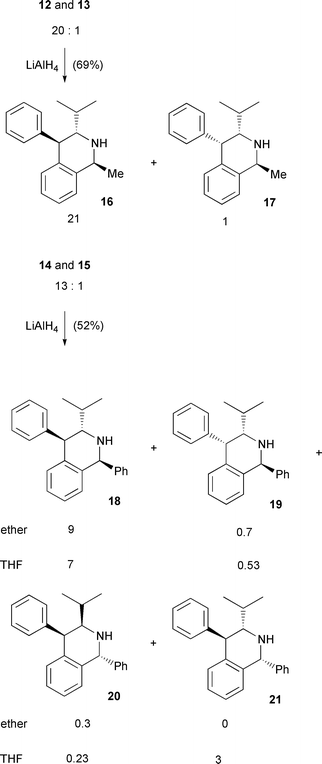 | ||
| Scheme 3 | ||
In a recent report5 the cyclisation of enantiomerically pure substrates 22 and 24 followed by in situ reduction of the 4-substituted N-methyldihydroisoquinolines with NaBH4 resulted in products with diastereoisomeric ratios ranging from 40–90% de as shown in Scheme 4. In that study the major products 23 and 25 have the C(1) and C(4) substituents cis to each other, similar to those observed in this study. For the reactions illustrated in Scheme 3, reduction of the Bischler–Napieralski generated tetrahydroisoquinolines 12–15 was highly stereoselective, generating a substantial preference for C(1)–C(4) cis products. Additionally the products have a trans relationship between the C(1) and C(3) substituents. Clearly the steric interactions arising from the C(3) and C(4) substituents are both reinforcing in the current study and contribute to the extremely high stereoselectivity observed for the reductions in Scheme 3.
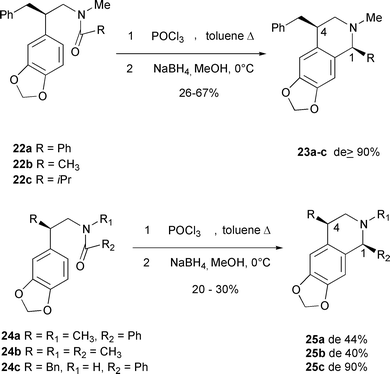 | ||
| Scheme 4 Related reductions of Bischler–Napieralski tetrahydroisoquinoline products showing a preference for C(1)–C(4) cis products.5 | ||
In summary, the amides of the amines 5 and 6 have been used to explore an asymmetric Bischler–Napieralski reaction. The cyclisation occurs with good stereoselectivity (80–90% de) to deliver 3,4-dihydroisoquinolines containing two contiguous stereogenic centres at C(3) and C(4). Further reduction of these products with LiAlH4 generates tetrahydroisoquinolines in a completely stereoselective manner, with a trans geometry between the substituents at C(1) and C(3) and a preferred cis geometry between the substituents at C(1) and C(4), to generate tetrahydroisoquinolines with three stereogenic centres.
Experimental
A GallenKamp GRIFFIN MPA350.BM2.5 melting point apparatus was used to record melting points, which are uncorrected. High-resolution mass spectrometry was performed on a VG AUTOSPEC spectrometer. 1H- and 13C-NMR spectra were recorded on a Varian Gemini 300 MHz spectrometer (1H at 299.98 MHz, 13C at 75.431 MHz). 1H-NMR NOE experiments were conducted on a Varian Unity Plus 500 MHz spectrometer (1H at 500.08 MHz). Chiral HPLC was carried out on a Chiralcel OD-H column with a Varian 9012 HPLC pump and Varian 9012 UV detector. All solvents were dried prior to use and reactions were carried out under an atmosphere of N2.(S)-N-(1-Methyl-2,2-diphenylethyl)acetamide 7
Triethylamine (0.34 ml, 2.41 mmol) and acetyl chloride (0.13 ml, 1.77 mmol) were added dropwise to a solution of amine 5 (340 mg, 1.61 mmol) in DCM (15 ml) at 0 °C. The reaction was stirred at 25 °C for 12 h and was then worked up by washing with water (3 × 20 ml). The organic layer was dried (MgSO4), filtered and evaporated under reduced pressure and the residue was purified by recrystallisation (petrol–acetone 90 ∶ 10) to give the product amide 7 as a colourless crystalline salt (389 mg, 94%). Mp 104–106 °C. For spectroscopic data see Table 2 (Found MH+, 254.1545. C17H19NO requires MH 254.1551).| Product | IR (ν/cm−1) | [α]20D/10−1 deg cm2 g−1 | δ | MS m/z (rel. int. %) | |
|---|---|---|---|---|---|
| 1H NMR (CDCl3, 300 MHz) | 13C NMR (CDCl3, 75.43 MHz) | ||||
| 7 | (KBr) 3445, 1546 (N–H), 1639 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
−67.2
(c = 0.64, CHCl3) |
1.1 (d, 3H, J = 6.4 Hz, CH–CH3), 1.73 (s, 3H, CO–CH3), 3.8 (d, 1H, J = 9.8 Hz, Ph2–CH), 4.8 (m, 1H, NH–CH–CH3), 5.18 (1H, d broad, J = 8.3 Hz, NH D2O exchangeable), 7.1–7.22 (m, 10H, aromatics) | 20.4 (CH3), 23.4 (CH3), 47.5 (CH–Ph2), 58.1 (CH–NH), 126.6, 128.11, 128.2, 128.5, 128.6, 141.8, 142.2 (aromatics), 169.0 (CO) |
(CI-CH4) 254 (MH+) (100%), 86 (Me–CH–NH–CO–CH3) (5%) |
| 8 | (KBr) 3407, 1556 (N–H), 1646 (CO) |
−35.3
(c = 0.56 CHCl3) |
0.99 (3H, d, J = 6.7 Hz, CH–CH3), 1.1 (3H, d, J = 6.7 Hz, CH–CH3), 1.8 (3H + 1H, m, CH3–CH–CH3 + CO–CH3), 4.01 (1H, d, J = 10.6 Hz, Ph2–CH), 4.99 (1H, m, NH–CH–iPr), 5.18 (1H, d broad, J = 9.1 Hz, NH D2O exchangeable), 7.2–7.5 (10 H, m, aromatics ) | 15.5 (CH3), 21.2 (CH3), 23.6 (CH3–CO), 29.3 (CH3–CH–CH3), 55.6 (CH–NH), 55.7 (Ph–CH–Ph), 126.8, 126.9, 128.2, 128.5, 128.8, 129.2 142.8, 143.0 (aromatics), 170.4 (CO) |
(CI-CH4) 282 (MH+) (100%), 114 (iPr–CH–NH–CO–CH3) (20%) |
| 9 | (KBr) 3341, 1532, (N–H), 1635 (CO) |
−29.5
(c = 0.59, CHCl3) |
1.0 (3H, d, J = 6.7 Hz, CH–CH3), 1.1 (3H, d, J = 6.7 Hz, CH–CH3), 1.95 (1H, m, CH3–CH–CH3), 4.2 (1H, d, J = 11.1 Hz, Ph2–CH), 5.2 (1H, m, NH–CH–iPr), 5.8 (1H, d broad, J = 10.1 Hz, NH D2O exchangeable), 7.1–7.6 (15H, m, aromatics H) | 15.5 (CH3), 20.9 (CH3), 29.3 (CH3–CH–CH3), 55.3 (CH–NH), 55.9 (Ph–CH–Ph), 126.5, 126.6, 127.8, 128.0, 128.3, 128.6, 130.9 142.3, 142.5 (aromatics), 168 (CO) |
(CI-CH4) 344 (MH+) (100%), 176 (Ph–CO+H–NH–CH–iPr) (15%), 75 (iPr–CH–NH) (26%) |
| 10 | (NaCl) 1631 (CN) |
— | 1.4 (3H, d, J
= 6.7 Hz, CH–CH3), 2.59 (3H, d, J
= 1.9 Hz, NC–CH3), 3.8 (1H, d, J
= 11.1 Hz, Ph2–CH), 3.9 (1H, m, N–CH–CH3), 6.9 (1H, m, aromatic), 7.2–7.5 (7H, m, aromatics H), 7.6 (1H, m, aromatic) |
21.8 (CH3), 23.4 (CH3), 49.5 (Ph–CH–ring), 57.7 (CH–N), 126.5, 125.2, 126.8, 127.8, 128.6, 129.0, 140.2, 141.9 (aromatics), 163.3 (CN) |
(CI-CH4) 236 (MH+) (100%) |
| 12 | (KBr) 1626 (CN) |
— | 1.05 (3H, d, J
= 6.7 Hz, CH–CH3), 1.20 (3H, d, J
= 6.7 Hz, CH–CH3), 1.85 (1H, m, CH3–HC–CH3), 2.6 (3H, d, J
= 1.5 Hz, NC–CH3), 4.0 (1H, dd, J
= 8.7, J
= 4.8 Hz, N–CH–CH–Ph), 4.2 (1H, d, J
= 9.7 Hz, N–CH–CH–Ph), 6.85 (1H, m, aromatic), 7.15–7.35 (7H, m, aromatics H), 7.58 (1H, m, aromatic) |
17.8 (CH3), 20.9 (CH3), 23.8 (CH3–CN), 30.9 (CH3–CH–CH3), 45.3 (Ph–CH–ring), 67.8 (CH–N), 125.2, 126.9, 127.2, 128.4, 128.9, 129.1, 129.5, 131.0, 140.5, 143.2 (aromatics), 162.9 (CN) |
(EI) 263 (M+) (44.5%), 248 (M+ − CH3) (100%), 220 (M+ − iPr) (220) |
| 14 | (KBr) 1608 (CN) |
— | 1.06 (3H, d, J = 6.7 Hz, CH–CH3), 1.20 (3H, d, J = 6.7 Hz, CH–CH3), 1.82 (1H, m, CH3–HC–CH3), 3.84 (1H, dd, J = 8.7, J = 4.5 Hz, N–CH–CH–Ph), 4.0 (1H, d, J = 8.7 Hz, N–CH–CH–Ph), 6.85–7.63 (14H, m, aromatic H) | 17.9 (CH3), 20.5 (CH3), 21.8 (CH3–CN), 30.2 (CH3–CH–CH3), 44.9 (Ph–CH–Ph), 67.9 (CH–N), 124.2–129.0, 141.2, 142.3 (aromatics), 165.4 (CN) |
(CI-CH4) 326 (MH+) (100%) |
| 16 | — | — | 0.98 (3H, d, J = 7.1 Hz, CH–CH3), 1.01 (3H, d, J = 7.1 Hz, CH–CH3), 1.56 (3H, d, J = 6.6 Hz, NH–CH–CH3), 1.7 (1H, m, CH3–HC–CH3), 2.97 (1H dd, J = 6.2, J = 6.2 Hz, NH–CH–CH–Ph), 4.0 (1H, d, J = 7.1 Hz, NH–CH–CH–Ph), 4.3 (1H, q, J = 6.6 Hz, CH–CH3), 6.80–7.3 (9H m aromatics) | 16.7 (CH3), 20.8 (CH3), 23.5 (CH3–CH–NH), 27.7 (CH3–CH–CH3), 47.9 (NH–CH–CH–Ph), 49.4 (–ring–CH–Me), 61.1 (NH–CH–CH–Ph), 128.2–130.4, 137.3, 140.7, 145.2 (aromatics) | (CI-CH4) 266 (MH+) (100%); 222 (MH+ − NH–CH–CH3) (22%) |
| 18 | — | — | 0.8 (3H, d, J = 6.8 Hz, CH–CH3), 1.01 (3H, d, J = 6.8 Hz, CH–CH3), 1.67 (1H, m, CH3–HC–CH3), 2.84 (1H, dd, J = 7.9, J = 4.9 Hz, NH–CH–CH–Ph), 4.1 (1H, d, J = 7.9 Hz, NH–CH–CH–Ph), 5.3 (1H, s, CH–Ph), 6.90–7.45 (14 H, m, aromatic) | 16.3 (CH3), 20.6 (CH3), 27.7 (CH3–CH–CH3), 47.9 (NH–CH–CH–Ph), 58.9 (NH–CH–ring), 61.1 (NH–CH–CH–Ph), 128.2–130.4, 137.1, 139.2, 144.9, 145.2, 140.7, 145.2 (aromatics) | (CI-CH4) 328 (MH+) (100%) |
(S)-N-(1-Benzhydryl-2-methylpropyl)acetamide 8
Triethylamine (0.88 ml, 6.27 mmol) and acetyl chloride (0.33 ml, 4.60 mmol) were added dropwise to a solution of amine 7 (1 g, 4.18 mmol) in DCM (40 ml) 0 °C and the mixture was then heated under reflux for 4 h. The reaction was washed with water (3 × 30 ml) and the organic layer was dried (MgSO4) and evaporated under reduced pressure to leave an amorphous powder. This material was recrystallised from hexane–ethyl acetate (85 ∶ 15) to obtain the title amide 8 as a colourless crystalline solid (1.10 g, 94%). Mp 130–132 °C. For spectroscopic data see Table 2 (Found MH+, 282.1858. C19H23NO requires MH 282.1850).(S)-N-(1-Benzhydryl-2-methylpropyl)benzamide 9
Triethylamine (0.40 ml, 2.19 mmol) and acetyl chloride (0.26 ml, 1.61 mmol) were added to a solution of amine 6 (350 mg, 1.46 mmol) in DCM (20 ml) at 0 °C and the reaction was stirred for 12 h at 25 °C. The reaction mixture was washed with water (3 × 20 ml) and the organic layer was dried (MgSO4), filtered and evaporated under reduced pressure. The product was recrystallised from ethanol–water (80 ∶ 20) to obtain the title amide 9 as a colourless crystalline solid (410 mg, 94%). Mp 221–223 °C (Found MH+, 344.2009. C24H26N1O requires MH, 344.2014). For spectroscopic data see Table 2.(3S,4S)-1,3-Dimethyl-4-phenyl-3,4-dihydroisoquinoline 10 and (3S,4R)-1,3-dimethyl-4-phenyl-3,4-dihydroisoquinoline 11
Phosphoryl chloride (1.35 g, 9.4 mmol) was added dropwise to a suspension of phosphorus pentoxide (0.86 ml, 9.4 mmol) in a solution of amide 7 (200 mg, 0.94 mmol) in anhydrous toluene (25 ml). After complete addition the reaction mixture was heated under reflux for 12 hours and then crushed ice (25 g) was added. The organic layer was separated and the aqueous residue was made basic with sodium hydroxide (15% w/v) solution and the product extracted into chloroform (3 × 25ml). The chloroform extract was dried (MgSO4), evaporated under reduced pressure and the product purified over silica (50 ∶ 50 hexane–ethyl acetate) to give 10–11 as a yellow oil (47 mg, 29%) (Found MH+, 236.1444. C17H18N1 requires MH, 236.1431). For spectroscopic data see Table 2.(3S,4S)-1-Methyl-3-isopropyl-4-phenyl-3,4-dihydroisoquinoline 12 and (3S,4R)-1-methyl-3-isopropyl-4-phenyl-3,4-dihydroisoquinoline 13
Phosphoryl chloride (3.25 ml, 35 mmol) was added dropwise to a suspension of phosphorus pentoxide (5.0 g, 35 mmol) in a solution of amide 8 (1.0 g, 3.54 mmol) in anhydrous toluene (60 ml). After complete addition the reaction mixture was heated under reflux for 12 hours and then crushed ice (60 g) was added. The organic layer was separated, the aqueous residue was made basic with sodium hydroxide (15% w/v) solution and the product was then extracted into chloroform (3 × 25ml). The chloroform extract was dried (MgSO4), evaporated under reduced pressure and the product purified over silica (95 ∶ 5 petrol–acetone) to give 12–13 as a colourless amorphous solid (558 mg, 60%) (Found M+, 263.1666. C19H21N1 requires M, 263.1674). For spectroscopic data see Table 2.(3S,4S)-3-Isopropyl-1,4-diphenyl-3,4-dihydroisoquinoline 14 and (3S,4R)-3-isopropyl-1,4-diphenyl-3,4-dihydroisoquinoline 15
Phosphoryl chloride (1.24 g, 8.9 mmol) was added dropwise to a suspension of phosphorus pentoxide (0.80 ml, 8.9 mmol) in a solution of amide 9 (303 mg, 0.88 mmol) in anhydrous toluene (25 ml). After complete addition the reaction mixture was heated under reflux for 12 hours and was then quenched by the addition of crushed ice (25 ml). The aqueous residue was made basic by the addition of sodium hydroxide (15% w/v) solution and the product was then extracted into chloroform (3 × 25ml). The chloroform extract was dried (MgSO4), evaporated under reduced pressure and the product purified over silica (90 ∶ 10 hexane–ethyl acetate) to give 14–15 as a colourless crystalline solid (91 mg, 32%) (Found MH+, 326.1915. C24H23N requires MH, 326.1908). For spectroscopic data see Table 2.(1S,3S,4S)-1-Methyl-3-isopropyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline 16 and (1S,3S,4R)-1-methyl-3-isopropyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline 17
A solution of 14–15 (352 mg, 1.33 mmol) in ether (30 ml) was added dropwise at 0 °C to a suspension of LiAlH4 (504 mg, 13.3 mmol) in ether (10 ml) and the reaction was heated under refluxed for 24 h. The reaction was then quenched with 5% HCl, the aqueous layer made basic with 15% NaOH and the products extracted into ether (3 × 30 ml). The organic extract was dried (MgSO4) and evaporated under reduced pressure to give 16 and 17 as a colourless oil (242 mg, 69 %) (Found MH+, 266.1901. C19H24N requires MH, 266.1908). For spectroscopic data see Table 2.(1S,3S,4S)-3-Isopropyl-1,4-diphenyl-1,2,3,4-tetrahydroisoquinoline 18, (1S,3S,4R)-3-isopropyl-1,4-diphenyl-1,2,3,4-tetrahydroisoquinoline 19 and (1R,3R,4S)-3-isopropyl-1,4-diphenyl-1,2,3,4-tetrahydroisoquinoline 20
A solution of 16–17 (48 mg, 0.147 mmol) in ether (5 ml) was added dropwise at 0 °C to a suspension of LiAlH4 (56 mg, 1.47 mmol) in ether (5 ml). The reaction was heated under reflux for 24 h and was then quenched with 5% HCl. The aqueous layer was made basic with 15% NaOH and the products were extracted into ether (3 × 10 ml). The organic extract was dried (MgSO4) and evaporated under reduced pressure to give 18 (and 19–20) as a colourless oil (25 mg, 52%) (Found MH+, 328.2058. C24H26N requires MH, 328.2065). For spectroscopic data see Table 2.Crystal structure determination of 12 and 14†
Data for both compounds were measured on a Bruker SMART diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.7107 Å) using 0.3° width steps accumulating area detector frames spanning a hemisphere of reciprocal space for both structures; the reflections were corrected for Lorentz and polarisation effects. Absorption effects were corrected on the basis of multiple equivalent reflections.The structures were solved by direct methods and refined by full matrix least squares on F2 using the program SHELXTL. All hydrogen atoms were included in calculated positions using a riding model. All non-hydrogen atoms were refined as anisotropic.
Acknowledgements
We thank the European Commission for supporting a Studentship (MN) through Research Training Network, ERBFMRXCT9, and we thank Onyx Scientific Ltd, Sunderland, UK, for a generous gift of amines 5 and 6.References
- A. Bischler and B. Napieralski, Ber. Dtsch. Chem. Ges., 1893, 26, 1903 Search PubMed.
- S. Nagubandi and G. Fodor, Heterocycl. Chem., 1980, 17, 1457 Search PubMed.
- G. Fodor, J. Gal and B. A. Phillips, Angew. Chem., Int. Ed. Engl., 1972, 11, 919 CrossRef CAS.
- M. D. Rozwadowska, Heterocycles, 1994, 39, 903 Search PubMed.
- V. Jullian, J.-C. Quirion and H.-P. Hussion, Eur. J. Org. Chem., 2000, 1319 CrossRef CAS.
- V. Vecchietti, G. D. Clarke, R. Colle, G. Dondio, G. Giardina, G. Petrone and M. Sbacchi, J. Med. Chem., 1992, 35, 2970 CrossRef CAS.
- T. Ishikawa, K. Shimooka, T. Narioka, S. Noguchi, T. Saito, A. Ishikawa, E. Yamazaki, T. Harayama, H. Seki and K. Yamaguchi, J. Org. Chem., 2000, 65, 9143 CrossRef CAS.
- D. O'Hagan and M. Tavasli, Tetrahedron: Asymmetry, 1999, 10, 1189 CrossRef CAS.
- F. Sanchez-Sancho, E. Mann and B. Herradón, Synlett, 2000, 4, 509.
Footnote |
| † CCDC reference numbers 168569 and 168570. See http://www.rsc.org/suppdata/p1/b1/b106942j/ for crystallographic files in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2002 |