Benzocycloarene hydroxylation by P450 biocatalysis
Martin P. Mayhew, Adrian E. Roitberg†, Yadu Tewari, Marcia J. Holden, David J. Vanderah and Vincent L. Vilker*
Biotechnology Division, National Institute of Standards and Technology, Gaithersburg, MD, 20899, USA. E-mail: vilker@nist.gov
First published on 8th January 2002
Abstract
Experimental and theoretical studies of the hydroxylation of a family of benzocycloarene compounds [benzocyclobutene, benzocyclopentene (indan), benzocyclohexene (tetralin), and benzocycloheptene] by wild type and Y96F mutant P450cam were performed in order to understand the factors affecting product distribution, catalytic rate and cofactor utilization. The products of all reactions except that of benzocycloheptene were regiospecifically hydroxylated in the 1-position. Reaction energetics predominated over active site steric constraints in this case so that quantum mechanical calculations (B3LYP/6-31G*) comparing the energetics of all possible radical intermediates successfully predicted hydroxylation at the 1- and 3-positions of benzocycloheptene, and at the 1-position for the other three compounds. However, the fact that the ratio of 1-alcohol to 3-alcohol changes significantly between wild type and Y96F mutant P450cam indicates that active site geometry and composition also play a significant role in determining BCA7 product regiospecificity. The indan and tetralin reaction products were stereoselective for the R enantiomer (88 and 94%, respectively). Steric constraints of the active site were confirmed by molecular dynamics calculations (locally enhanced sampling dynamics) to control enantiomer distribution for tetralin hydroxylation. NADH coupling, binding affinity, and product turnover rates were dramatically higher for Y96F P450cam, showing that the removal of the active site hydroxyl group on tyrosine makes the enzyme better suited for oxidation of these hydrophobic compounds. NADH coupling, binding affinity and product turnover rate for each enzyme generally increased with arene ring size. For both enzymes, NADH coupling and product turnover rates were correlated with the extent of high-spin shift upon substrate binding as determined by the shift in Soret absorption bands at 417 and 391 nm.
Oxygenated benzocycloarenes (fused benzene–cycloalkane structures) are useful reactants in asymmetric syntheses.1 Toluene and naphthalene dioxygenases, and cytochrome P450 monooxygenases, either in the context of whole cells or reconstituted enzyme systems, have been investigated as biocatalysts for hydroxylation of benzocycloarenes to regio- and stereospecific alcohols.2–6 Considered as a suite of catalysts, these three enzymes offer many different possibilities for the enzymatic insertion of the hydroxyl functionality, including mono-ol or di-ol, on the benzene ring and for several monooxygenation positions on the fused aliphatic ring. Cytochrome P450 catalyzes only monooxygenation on the aliphatic ring. In part, the present work differentiates the catalytic functionality of one P450 monooxygenase from the dioxygenases that have been applied to this class of substrates.
The cytochrome P450 superfamily of enzymes is ubiquitous in nature, existing in a wide variety of organisms, from bacteria to humans.7 For the potential impact of P450 biocatalysis in industrial fine chemical synthesis to become a reality, methods to identify potential catalytic target compounds and to improve natural P450 enzymes for use with non-natural substrates of interest must be developed. Slow reaction rates and cofactor utilization efficiencies for non-natural substrates hinder the application of P450 enzymology to large-scale chemical syntheses.
Numerous studies have explored ways to improve P450s as effective biocatalysts. Through site-directed mutagenesis, active-site residues critical to substrate specificity, product regiospecificity, and rates of catalysis have been elucidated for many P450s, including P450cam,8 P450 BM3,9 P450 1A2,10 and P450 2B4.11 P450 enzymes with enhanced stability and solubility have been made using site-directed mutagenesis and truncation of hydrophobic tails,12–14 and creation of a bacterial–human P450 chimera.15 A thermostable P450, CYP119, has been isolated from the archaeon Sulfolobus sulfataricus16 and its biocatalytic properties and structure determined.17,18
Several studies have used computer-based algorithms to investigate the predictability of ligand–P450 interactions.19–24 In early work, the active site of the enzyme was kept rigid, but later more refined calculations included full dynamical properties of the system. Loew's group in particular pioneered the use of joint dynamical/energetic criteria for prediction and explanation of observed hydroxylation trends.19,21 These criteria were then used by Loew and others to successfully explain trends in hydroxylation of camphor, camphane, thiocamphor and norcamphor.19,23,24 There is a need for additional systemic experimental information in order to model the pertinent P450 structural and dynamic features that allow for prediction of novel P450 catalytic activities. This need is in part due to the fact that there are very few known structures of P450 enzymes when compared to the number of P450s of immediate scientific and industrial interest. Presently, these less well-known P450s are investigated using sequence homology models to enzymes with known structure.25
This study focuses on P450cam (CYP101), the three-protein enzyme system that metabolizes camphor in Pseudomonas putida
(ATCC 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 453), and the mutant Y96F P450cam for which the active site pocket is more hydrophobic. The binding of the native substrate, camphor, to P450cam acts as a redox gate for the enzyme system, as the redox potential of the enzyme shifts to enable electron acceptance from putidaredoxin. The coupling of NADH oxidation to camphor hydroxylation is nearly 100%. For non-natural substrates, however, NADH coupling and turnover rate decrease, as formation of hydrogen peroxide, superoxide, or water precedes catalytic turnover.26 Increased water access to the reduced, non-native substrate-bound active site is believed to cause less efficient NADH coupling and slower reaction rates. The removal of the active site hydroxyl group upon replacement of tyrosine 96 with phenylalanine has been shown to improve
P450cam catalysis of hydrophobic substrates such as styrene,8,27 alkanes,28 and polycyclic aromatic hydrocarbons.29
453), and the mutant Y96F P450cam for which the active site pocket is more hydrophobic. The binding of the native substrate, camphor, to P450cam acts as a redox gate for the enzyme system, as the redox potential of the enzyme shifts to enable electron acceptance from putidaredoxin. The coupling of NADH oxidation to camphor hydroxylation is nearly 100%. For non-natural substrates, however, NADH coupling and turnover rate decrease, as formation of hydrogen peroxide, superoxide, or water precedes catalytic turnover.26 Increased water access to the reduced, non-native substrate-bound active site is believed to cause less efficient NADH coupling and slower reaction rates. The removal of the active site hydroxyl group upon replacement of tyrosine 96 with phenylalanine has been shown to improve
P450cam catalysis of hydrophobic substrates such as styrene,8,27 alkanes,28 and polycyclic aromatic hydrocarbons.29
In this work, we studied the biocatalysis of wild type and the Y96F mutant P450cam toward four benzocycloarene compounds of increasing aliphatic ring carbon number: benzocyclobutene, benzocyclopentene (indan), benzocyclohexene (tetralin) and benzocycloheptene. Products were identified, and kinetic parameters and NADH coupling efficiencies determined for each substrate. Regiospecific product distributions were determined, with stereochemistry determined for the indan and tetralin reaction products. A theoretical protocol employing both dynamic and thermodynamic criteria was used to predict the regio- and stereospecificity of these reactions. A spectrophotometric assay that measures the Soret band shift upon substrate binding was developed and compared with NADH coupling and product turnover rates to assess this assay's potential as a biocatalyst screening tool.
Materials and methods‡
Principal substances
Most substances used in this study were purchased commercially at their highest level of purity and used without further purification. NADH (CAS 606-68-8), dimethyl sulfoxide (DMSO; CAS 67-68-5), benzocyclobutene (BCA4; 1,2-dihydrobenzocyclobutene; CAS 694-87-1), benzocyclopentene (BCA5; indan; CAS 496-11-7), and benzocyclohexene (BCA6; 1,2,3,4-tetrahydronaphthalene; tetralin; CAS 119-64-2) were purchased from Sigma-Aldrich (St. Louis, MO) with purities of 98, 99.5, 99, 95, and 99 wt%, respectively. Reported purities of the benzocycloarene compounds were confirmed by in-house GC analysis. Benzocyclohepten-3-ol was kindly provided by Dr Derek R. Boyd and the structure was confirmed by GC-MS.Protein preparation and absorbance measurements
Protein expression, site-directed mutagenesis, and purification of putidaredoxin (Pdx), putidaredoxin reductase (PdR), and cytochrome P450cam were performed as previously described.8 Prior to incubation with substrates, proteins were buffer-exchanged with Buffer T (50 mM TRIS-HCl, 0.2 mM KCl, pH 7.4 at 22°C) by passing through Sephadex G-25 superfine desalting resin (Amersham-Pharmacia Biotech, Piscataway, NJ) at 4°C. Wild type (WT) and mutant P450cam proteins were treated as follows to ensure complete removal of camphor. P450cam samples were made 50 mM in dithiothreitol (DTT; Diagnostic Chemicals, Ltd., Oxford, CT) and incubated at 22°C for 15 min. Next, the proteins were buffer exchanged at 4°C with 10 mM TRIS-HCl, 1 mM DTT, pH 7.4 at 22°C using desalting resin. Protein that eluted from the column was concentrated using either a Centricon-10
or Centriprep-10 centrifugal concentrator (Millipore, Inc., Bedford, MA). P450cam proteins were then passed through another Sephadex G-25 superfine column that was equilibrated with buffer T at 4°C. Protein concentrations were quantified from absorption spectra using Pdx, ε455=10.4 mM−1 cm−1; PdR, ε454=8.50 mM−1 cm−1; for P450cam devoid of camphor at 25°C, ε417,WT=112 mM−1 cm−1 and ε417,Y96F=107 mM−1 cm−1. These data come from Gunsalus and Wagner,30 and recently performed measurements from our laboratory for the Y96F mutant.8The percentage of high-spin state iron in solutions of P450cam, with and without benzocycloarene substrates, was determined by a spectrophotometric method.31 Solutions of 100 mM DMSO (see Reaction kinetics and NADH coupling measurements) and various concentrations of benzocycloarene in buffer T were allowed to equilibrate in a stirred, thermostatic spectrophotometer setup (25°C, 2 min equilibration). Wild type or Y96F P450cam (5 μM) was added and spectra from 350 to 600 nm were recorded after an additional 2 min equilibration period. The same experiment was performed with P450cam solutions devoid of substrate, and solutions with 0.2 mM camphor added. In each case, various aspects of the spectra, most notably the isobestic point at 405 nm, were examined to ensure consistent quality of the spectra.
Syntheses
°C. After removal of the aluminum foil the temperature was stabilized at ∼225°C and held for an additional 2 h. After cooling, the reaction mixture was diluted with H2O and the product was then extracted into 95% hexanes (2×100 mL). The combined hexane extracts were washed with brine, dried (MgSO4), and concentrated under reduced pressure to give 4.32 g (94.7%) of a liquid product.
Flash column chromatography yielded 3.71 g of pure product. The purity of the compound was checked by gas chromatography and its structure was confirmed by IR, proton NMR, and GC-MS.×100 mL). The combined methylene chloride extracts were washed with brine, dried over magnesium sulfate, and concentrated to give ∼6.0 g of crude solid product. Recrystallization from ∼50 mL of 95% hexanes yielded 3.84 g of purified product. The melting point of the synthesized benzocyclohepten-1-ol was measured to be 100–101.7°C, compared to the reported value32
of 100–101°C. The benzocyclohepten-1-ol structure was confirmed by proton NMR and purity was checked by gas chromatography.Extraction and measurement of reaction products
All reactions in which oxidized products were to be measured were performed in 1.0 mL glass septum microvials (Kimble, Vineland, NJ). Extraction was performed in the same microvial as the reaction in order to reduce systematic and experimental errors. Internal standards were n-decane (Sigma–Aldrich, St. Louis, MO) for analysis of BCA4 and BCA5 in reaction extracts, BCA7 for analysis of BCA6 in reaction extracts, and BCA6 for analysis of BCA7 in reaction extracts. In all cases, the enzyme reaction was quenched by addition of chloroform. After vigorous agitation followed by centrifugation, aliquots (1.0 μL) of the chloroform phase were injected into an HP 5890 GC chromatograph fitted with a packed column injector (modified for use with capillary columns) and an FID detector. Different GC columns and temperature settings were used for each substrate to optimize resolution and sensitivity. All analytes were baseline-resolved. BCA4 and BCA5 reaction extracts were analyzed using an HP-5 cross-linked phenylmethyl silicone glass capillary column (30 m×0.53 mm×0.88 μm film thickness; Hewlett–Packard, Wilmington, DE). BCA6 and BCA7 reaction extracts were analyzed with a ZB-WAX polyethylene glycol glass capillary column (30 m×0.53 mm×1.0 μm film thickness; Phenomenex, Inc., Torrance, CA). Chromatographic response factors were measured for all substrates, products, and internal standards using authentic samples, except for benzocyclobuten-1-ol, which was assumed to have the same response factor as that of BCA4, and benzocyclobuten-3-ol, which was assumed to have that of benzocyclobuten-1-ol.Oxidized product identification was done by GC-MS analysis (SAIC, Inc., NCI-FCRDC, Frederick, MD). Electron impact source energy was 70 eV, and GC conditions were similar to the conditions stated above. Both un-derivatized and TMS-derivatized samples of each benzocycloarene reaction extract were analyzed by GC-MS. In each case, mass spectra for reaction extracts were compared with mass spectra from an authentic sample of the putative reaction product. Benzocyclobuten-1-ol was identified by analysis of the derivatized and un-derivatized mass spectra of BCA4 reaction extracts.
The enantiomeric excess of the hydroxylated products from BCA5 and BCA6 hydroxylation were analyzed by chiral HPLC. Poor separation of the BCA4 and BCA7 hydroxylation products prevented the establishment of the enantiomeric purity of these compounds. Hexane extracts of reaction solutions were analyzed on an HP Series 1100 HPLC, fitted with a Chiralcel OB-H cellulose column (46 mm×5 mm; Chiral Technologies, Exton, PA). The isocratic separation was performed in 5% isopropyl alcohol–95% hexane, at 0.5 mL min−1. Compounds were detected by monitoring absorbance at 254 nm. The retention times were: BCA5 (7.7 min), BCA6 (7.8 min), (R)-1-tetralol (13.7 min), (R)-1-indanol (16.5 min), (S)-1-tetralol (26 min), (S)-1-indanol (29 min). Due to a lack of authentic enantiopure 1-indanol, it was assumed
that the R enantiomer eluted first by analogy with (R)-1-tetralol.
Saturation molality of benzocycloarene compounds
The saturation molality of each benzocycloarene was determined by approaching equilibrium from two different temperatures. Approximately 0.02 g of benzocycloarene was placed in two 50 ml Erlenmeyer flasks containing 40 g of Buffer T. One flask was placed in a shaker bath set at 15°C and the other in a shaker bath set at 37°C. After 24 h both flasks were placed in one shaker bath at 25°C and allowed to equilibrate for an additional 4 days. From each flask, 28 g of the equilibrated aqueous phase was transferred to a 40 ml Teflon tube, to which 1.3 g of n-hexane and 0.06 g of internal standard (1-decanol in hexane) were added. After vigorous agitation and centrifugation for 15 min at 3000 g, 1.0 μL of the hexane phase was injected into a Hewlett Packard (HP) 5890 gas chromatograph equipped with a flame ionization detector and a fused silica ZB-WAX column (30 m×0.53 mm×1.0 μm
film thickness; Phenomenex, Inc., Torrance, CA). Dichloromethane was used as the extractant instead of hexane in the benzocyclohepten-1-ol determination.Reaction kinetics and NADH coupling measurements
P450cam reaction kinetics for hydroxylation of the benzocycloarenes was determined by measuring the disappearance of NADH. The assay solution contained 10 μM Pdx, 1 μM PdR, 2000 U mL−1 catalase, 160 μM NADH, 100 mM DMSO, and varying concentrations of benzocycloarene. This DMSO co-solvent concentration was previously found to be optimal for BCA6 (tetralin) P450cam biocatalysis,6 although it had negligible effect on BCA6 solubility. No further attempts were made to optimize the concentration for the other enzyme-substrate pairs of this study. Assay solutions were placed into a quartz cuvette fitted with an electric stirrer (Spectracell, Oreland, PA), and were allowed to equilibrate for 2 min in a thermostatic cuvette holder (25°C). WT (1 μM) or Y96F (0.1 μM) P450cam was added to initiate the reaction, and the absorbance monitored at 340 nm to measure NADH disappearance (ε340=6.22
mM−1 cm−1). Pdx auto-oxidation, about 0.2 min−1 as measured in trials devoid of P450cam, was subtracted from the overall NADH utilization rate.Reactions for NADH coupling determinations were performed in 1.0 mL septum glass micro-vials containing 1 μM PdR, 2 μM Pdx, and 2000 U mL−1 bovine liver catalase (Sigma Chemical, St. Louis, MO) in Buffer T. Wild type P450cam reactions included 10–15 μM P450cam, whereas Y96F P450cam reactions included 2 μM P450cam. The benzocycloarenes were added as DMSO stock solutions to give solution concentrations of about 100 mM DMSO and near-saturation levels for each benzocycloarene. NADH at limiting concentrations (200–600 μM) was added to initiate the reaction. A low Pdx-to-P450cam ratio was used in order to minimize Pdx auto-oxidation. Reactions were complete within 5 min. Calibration solutions for quantifying oxidized products were made by adding known amounts of product to solutions that were identical to the reaction mixtures except that NAD+ was substituted for NADH. Calibration solutions of benzocyclobutene and benzocyclohepten-1-ol were used to estimate response factors for benzocyclobuten-1-ol and benzocyclohepten-3-ol, respectively. NADH coupling was calculated by dividing total product by the NADH added at the start of the reaction.
The rate of total hydroxylated products for each enzyme-substrate reaction was determined from NADH utilization rate times NADH coupling efficiency. This assumes constant proportionality between NADH utilization rate and substrate hydroxylation rate. The validity of this assumption will be addressed later. Michaelis–Menten parameters Km and kcat were obtained by nonlinear regressions (SigmaPlot 4.0, SPSS Science, Chicago, IL) of the total product rate determinations.
Theoretical calculation of reaction products
The molecular dynamics runs were performed using the program AMBER33 with the standard force field34 used for the P450cam protein. The force field for the heme group and the heme–thiolate bond, as well as the iron-oxo species were described by the parameters found in publications of Loew's group.35 BCA6 (tetralin) was selected as the model compound to represent binding and dynamical characteristics of all benzocycloarenes under investigation in this work. The program Leap36 was used to build the BCA6 molecule. A 6-31G** Hartree–Fock in vacuo calculation was done, and point charges assigned to classical atoms based on a global fit to the electrostatic potential.37 Equilibrium values for internal BCA6 degrees of freedom as well as force constants were assigned based on the same 6-31G** Hartree–Fock calculation. The initial structure used for the simulation came from the adamantane-bound P450cam crystal structure38 (Protein Data Bank ref. 5cpp), from which adamantane and all water molecules located more than 10 Å from the heme iron were removed. The bound conformation of BCA6 in the active site was estimated by initial placement of BCA6 in alignment (main axis of rotation) with the original adamantane, followed by use of the locally enhanced sampling (LES) method,39 as implemented in AMBER, to over-sample the ligand orientations within the heme pocket. Ten copies of the ligand were used, at a temperature of 150 K, for 200 ps. After the MD run, no correlation was found between the possible ligand positions, with extensive orientation randomization. A slow cooling protocol produced a single structure with all copies almost completely overlapped. This structure is designated as the BCA6 bound state. Also, MD runs with two very different (manually obtained) initial orientations of BCA6 inside the active site were made with a very detailed and slow equilibration method, similar to that used by Wade et al.40 With this method, the system was run for 40 ps at 150 K, with atomic harmonic restraints (AHR) using a force constant of 20 kJ mol−1 Å2. It was followed by 10 ps of dynamics at 300 K with AHR and a 20 kJ mol−1 Å2 force constant, 10 ps with AHR at 4 kJ mol−1 Å2, 10 ps at 0.4 kJ mol−1 Å2, and 10 ps of unrestrained dynamics. A production run of 1.5 ns was done for each of the initial structures. Both simulations converged not only to very similar structures, but also to a conformation very close to that obtained using the LES protocol.Tyrosine 96 was mutated to phenylalanine to give the mutant Y96F P450cam. A dielectric constant of 1 was used throughout the simulations. A spherical cutoff of 30 Å was used for the Lennard-Jones non-bonded potentials. The time step was 2 fs. The non-bonded list was updated every 20 fs. The bonds that involved an H atom were constrained to their equilibrium lengths by using the SHAKE method.
Structures for benzocyclopentene, benzocyclohexene, and benzocycloheptene were optimized with both semi-empirical (AM1 within Hyperchem) and density functional theory (B3LYP/6-31G*, within Gaussian 98)41 calculations. The energetics of the free radicals assumed to form during the reaction were computed at the same theory level, but under an unrestricted Hamiltonian, to account for the doublet spin state. The free radical energies were computed for the geometries of the original molecules, as well as for the locally minimized doublets.
Results and discussion
Benzocycloarene reaction products: measurements and comparisons with dioxygenases
Except for preliminary mass balance studies performed at sub-saturation concentrations and low substrate conversion, all reactions with WT and Y96F P450cam were carried out at saturation levels of benzocycloarene where the likelihood of generating secondary products from enzymatic attack on the primary hydroxylation products is minimized.6,8 Saturating solutions were made using the following solubility data acquired as part of this study [in the form quantity (standard error)]: BCA4=1580 (20)
μM, BCA5=690 (20)
μM, BCA6=359 (7)
μM, and BCA7=127 (6)
μM. The solubility obtained for BCA6 is in statistical agreement with our previous result of tetralin solubility as a function of buffer composition.42 Preliminary mass balance measurements made at sub-saturating
concentration of BCA5 and low substrate conversion (<20 mol%) indicated that only one product is formed during biocatalysis, similar to our earlier finding with styrene epoxidation by P450cam Y96F mutant.8Table 1 shows the products generated by WT or Y96F P450cam catalysis from each benzocycloarene. The 1-alcohol is the predominant reaction product in all cases. BCA7 is the only substrate that forms two regioisomers: benzocyclohepten-1-ol and benzocyclohepten-3-ol. The presence or absence of the active site tyrosine hydroxyl group is seen to affect the regioisomer product distribution for benzocycloheptene.
| Relative productsa | ||
|---|---|---|
| Substrate | Wild type | Y96F |
| a Wild type and Y96F benzocycloarene reaction products identified by GC-MS as described in Materials and methods. For the first three compounds, only one product was detected under the substrate saturating conditions used for the reactions.b Enantiomeric purity (±2%) indicated by numbers in parentheses (nd=not determined).c Numbers without parentheses indicate relative amount of products (±3%) found for BCA7 reaction products. | ||
 Benzocyclobutene (BCA4) Benzocyclobutene (BCA4) | 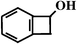 (nd)b (nd)b | 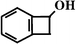 (nd) (nd) |
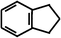 Benzocyclopentene (BCA5) Benzocyclopentene (BCA5) | 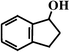 (87%
R) (87%
R) | 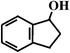 (87%
R) (87%
R) |
 Benzocyclohexene (BCA6) Benzocyclohexene (BCA6) | 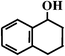 (95%
R) (95%
R) | 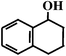 (93%
R) (93%
R) |
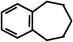 Benzocycloheptene (BCA7) Benzocycloheptene (BCA7) |  54%
(nd)c 54%
(nd)c | 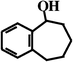 83%
(nd) 83%
(nd) |
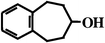 46%
(nd) 46%
(nd) | 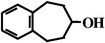 17%
(nd) 17%
(nd) | |
There are significant differences and similarities with the products formed by dioxygenase catalysis of these compounds. For biocatalysis using toluene dioxygenase (TDO): BCA4 is hydroxylated at multiple positions including attack on the benzyl ring3 while P450cam gives only the regioisomer 1-hydroxybenzocyclobutene; BCA5 is hydroxylated regio- and stereospecifically to 1-(R)-indanol, the same as for P450cam hydroxylation; BCA6 and BCA7 are not known substrates while P450cam forms a single regio- and stereospecific product [1-(R)-tetralol] from BCA6, and the 1-ol and 3-ol from BCA7. For biocatalysis by naphthalene dioxygenase (NDO): BCA4 is regiospecifically hydroxylated to the single product (±)-1-hydroxybenzocyclobutene,43 the same as for P450cam hydroxylation; BCA5 goes to multiple mono-ol and di-ol products but the major product is 1-(S)-indanol,2 opposite to the chirality given by TDO, and by P450cam where only one product is made; BCA6 is known not to be an NDO substrate43 and BCA7 is not a known substrate.
Benzocycloarene reaction products: theoretical calculations
Our calculations of the regio- and stereospecifity of P450 catalysis, largely based on the work of Harris and Loew,19,21,35 introduce both geometric and thermodynamic criteria using the assumption that P450cam hydroxylation is rate-limited by H-atom abstraction (and the formation of a free radical), with a subsequent, extremely fast rebound mechanism. The reactive intermediate at the heme is assumed to be compound I, radical cation ferryl oxygen. The geometric criterion was then extracted from a molecular dynamics (MD) run with appropriate parameters. The distances (and angles and torsions) between a number of H atoms in the substrate and the O atom in the ferryl oxygen heme species were monitored versus time, and a record was made of the number of close contacts seen during the dynamics run. The percentage of close approaches between the reactive species was assumed to correlate directly with the reaction probability at a given H site.Although Loew et al. have computed the energetics of free radicals after local optimization of the spin doublet,19 doubts persist as to the relevance of the calculation to the hydroxylation and hydrogen rebound. There are a number of publications dealing with the correct time scale for hydroxylation. Newcomb and Toy44 reviewed the use of hypersensitive radical probes used as radical clocks for P450 catalyzed hydroxylation reactions. One can see that the time scale for the reaction in the enzyme is somewhere in the nano- to picosecond range. Therefore, our calculations needed to consider if during that time the free radical generated during the H-atom abstraction has time to relax to its low energy conformation. We performed the following model calculation to decide on the time scale of the rearrangement: a semi-empirical molecular dynamics run was done with an AM1 Hamiltonian, starting from the geometry right after H abstraction, with no geometry relaxation. Following the change in time from sp3 to sp2 hybridization by monitoring the out-of-plane angle for the remaining H atom, allows us to estimate the time scale for rearrangement to be 40 fs. Even if this calculation is wrong by a factor of 100, the time scale for the reaction in vitro is quite comparable with the theoretical time scale for rearrangement. The comparison of energies for the un-optimized free radicals is not enough to provide a proper picture. Our results for both the optimized and un-optimized free radicals are shown in Table 2. Even these calculations can be misleading and should only be used for trends. A proper (but very expensive) calculation should account for differential energies inside the active site of the enzyme, and should provide for the possibility of free radical migration from the H-atom abstraction site to a neighboring carbon atom.
| Semiempirical AM1 | Density functional theory | ||||
|---|---|---|---|---|---|
| Ring size | Position | Un-optimized sp3 | Optimized sp2 | Un-optimized sp3 | Optimized sp2 |
| a Energies in kJ mol−1 are relative to lowest energy value (position 1) for the appropriate set. Non-optimized energies averaged over pro-R and pro-S hydrogen abstraction sites. | |||||
| 5 | 1 | 0 | 0 | 0 | 0 |
| 2 | 10.5 | 27.7 | 14.7 | 40.6 | |
| 6 | 1 | 0 | 0 | 0 | 0 |
| 2 | 17.6 | 35.6 | 21.4 | 45.5 | |
| 7 | 1 | 0 | 0 | 0 | 0 |
| 2 | 15.5 | 18.0 | 18.9 | 23.9 | |
| 3 | 2.1 | 4.6 | 3.8 | 6.3 | |
The results in Table 2 show that position 1 is always preferred for H-atom extraction, regardless of the ring size. The case of BCA7 (7-membered ring) is of particular interest. Table 2 shows that the energy difference between sites 1 and 3 is not as large as that as between sites 1 and 2. This effect arises from hyperconjugation between the C-3 free radical and the aromatic ring via the C-1 atom. An energy difference of 6 kJ mol−1 is small enough to rationalize a significant population of products hydroxylated in position 3. From the data in Table 2, it is predicted that the thermodynamic criteria presented above will favor 1-hydroxylations of BCA4, BCA5, and BCA6, and 1- and 3-hydroxylations of BCA7 upon biocatalysis with P450cam. The fact that the ratio of 1-alcohol to 3-alcohol changes significantly between wild type and Y96F P450cam indicates that active site geometry and composition also play a significant role in determining BCA7 product regiospecificity.
To provide the geometric criteria necessary for establishing active site effects on reaction regio- and stereospecificity, MD of a benzocycloarene compound in the P450cam active site was performed. First, the structure of P450cam bound with the benzocycloarenes had to be predicted. The result of these calculations is the bound conformation of BCA6 in Y96F P450cam as depicted in Fig. 1. In this conformation, BCA6 is in close contact (<3 Å) with 12 residues of the protein. The aromatic section is in contact with F87, F96 and F98, and the aliphatic region points towards the heme group. This picture supports the experimental fact that P450cam hydroxylates only the non-aromatic region of BCA6. Next, data from the dynamics was used to monitor distances from each non-aromatic carbon to the heme ferryl oxygen. Such an approach shows no discrimination between C-1 and C-2 for BCA6, with both atoms approaching the ferryl oxygen the same percentage
of the dynamics runs, within statistical bounds. Therefore, active site interactions place BCA6 (and likely the other benzocycloarenes) in a conformation amenable to non-aromatic hydroxylation, but do not provide a rational for determining regiospecificity on the non-aromatic portion of BCA6 (or smaller benzocycloarenes). Since BCA7 is larger than BCA6, it is not possible to infer active site effects on benzocycloheptene regio- and stereospecificity from the BCA6 MD runs.
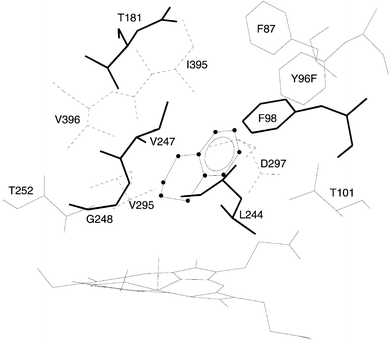 | ||
| Fig. 1 The bound state of benzocyclohexene (BCA6, tetralin) in the active site of Y96F P450cam. Line widths of the amino acid residues represent the third spatial dimension. | ||
We also computed the distances between the ferryl oxygen atom and the pro-R and pro-S hydrogen atoms at the C-1 position in BCA6 to predict whether BCA6 hydroxylation would be stereospecific. A histogram of such distances is shown in Fig. 2. These data are subject to uncertainties associated with not knowing the effects of high flexibility in the active site and in the choice of distance for defining a close encounter. Nevertheless, to a first approximation they suggest a significantly closer approach of the pro-R atom to the ferryl oxygen, and therefore that the pro-R hydrogen spends considerably more time close to the ferryl oxygen than does the pro-S hydrogen.
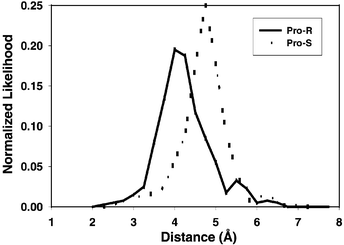 | ||
| Fig. 2 Histogram of the distances between the ferryl oxygen atom and the pro-R or pro-S hydrogen atom at the C-1 position in benzocyclohexene (BCA6, tetralin). Data compiled from a 1.5 ns molecular dynamics run. | ||
For dioxygenase enzymes, no theoretical studies of this type, or even simple MD, have been done. This is probably because only recently has a structure become available for NDO45 (there is no TDO structure yet published), and there is almost no mechanistic understanding of the dioxygenase catalytic cycle. As it is shown here, the regioselectivity of hydroxylation by P450cam can be rationalized by the relative energies of the free radicals formed in the H-rebound mechanism, along with steric constraints revealed through the MD simulations. Under this light, no possibility of hydroxylation on the aromatic ring exists. However, TDO seems to be able to hydroxylate BCA4 in both rings. A possible explanation includes a mechanism that does not have a free radical formed at any point. Based on the facts that the 5-membered aliphatic ring of BCA5 gives the same products with TDO and P450cam, that toluene is the natural TDO substrate, and that BCA6 and BCA7 are not TDO substrates, one can speculate that the TDO active site is too tight for the larger BCA molecules, while being too loose for BCA4, which can adopt multiple conformations in the active site and hence is subject to multiple reaction sites.
NADH coupling and kinetic characterizations
In order to study the effects of arene ring size and the Y96F mutation on P450cam biocatalysis, extensive kinetic analyses were performed on each enzyme-substrate reaction. The results are summarized in Table 3. While NADH coupling to WT P450cam is poor and uncorrelated with substrate size, coupling is markedly increased for Y96F P450cam on all of the benzocycloarenes. In addition, for the Y96F mutant, increasing the arene ring size also results in better NADH coupling. These trends are consistent with the hypothesis that poor NADH coupling arises from water access to the active site during catalysis.38 Removing the active site hydroxyl of tyrosine makes water access less energetically favorable, and the binding of larger substrates more effective blockers of heme iron electrophilic attack by water.| Wild type | Y96F | |||||
|---|---|---|---|---|---|---|
| Substrate | C (%) | Km/μM | kcat/s−1 | C (%) | Km/μM | kcat/s−1 |
| a NADH coupling (C), measured at saturating benzocycloarene and limiting NADH concentrations, calculated by dividing total product concentration after complete NADH exhaustion by initial NADH concentration. Michaelis–Menten parameters Km and kcat determined by nonlinear regression (SigmaPlot 4.0) of total product rates (product formation rate equals NADH utilization rate times NADH coupling efficiency). | ||||||
| BCA4 | 10±2 | 310±70 | 1.5±0.2 | 35±3 | 13±3 | 3.3±0.2 |
| BCA5 | 15±2 | 250±40 | 2.1±0.2 | 46±3 | 19±6 | 6.6±0.9 |
| BCA6 | 6±2 | 130±20 | 1.2±0.1 | 52±3 | 9.8±0.8 | 5.4±0.2 |
| BCA7 | 12±2 | 55±10 | 1.0±0.1 | 70±5 | 6±1 | 4.6±0.2 |
The kinetic characterization showed that Y96F is superior to WT P450cam enzyme in both substrate binding affinity and product turnover rate for all of the benzocycloarenes. The Km values for Y96F are an order of magnitude smaller than for WT, and kcat values are increased two- to fivefold, depending on the substrate. A clear trend of increasing binding affinity with increasing arene ring size is observed for WT, providing further evidence that larger ring sizes allow for a tighter fit into the active site. In addition, as suggested above, the absence of the active site hydroxyl in Y96F P450cam results in an order of magnitude greater binding affinity for each substrate. Why would the removal of the active site hydroxyl cause a reduced dependence of binding affinity on arene ring size? It could be rationalized that since both substrate and active site are almost completely hydrophobic in mutant Y96F, the increase in arene ring size promotes minor stabilization to binding. In contrast, for the WT enzyme, where substrates are stabilized by hydrophobic interactions within the active site cavity and destabilized by the active site hydroxyl, addition of more hydrophobic surface area to the substrate by increasing arene ring size provides a larger incremental stabilization.
Although product turnover rates (kcat) are larger for the Y96F mutant compared to WT, there is no correlation with substrate size for either enzyme. Binding affinity of a substrate has little correlation with its associated product turnover rate.26,46
Soret shift assay to predict P450 catalysis
All P450s are heme-thiolate proteins with a large Soret absorbance band at approximately 420 nm, corresponding to the predominant low-spin resting state. Upon substrate binding, the Soret band for most P450s is blue-shifted by about 20 nm, corresponding to a predominant high-spin form of the heme iron.31 This low-spin to high-spin transition adjusts the redox potential of P450 to allow for electron exchange with iron-sulfur or flavin protein redox partners, thereby modulating catalytic activity.47 Since the four benzocycloarenes tested in this study displayed a wide range of catalytic activity between the WT and Y96F mutant enzymes, we sought to learn if a Soret shift measurement could be developed that would correlate with these different catalytic activities. Turnover of native and non-native substrates by several different P450s has been reported along with Soret band absorbance data27,48 that indicate this might be an expedient optical method to screen for P450 activity. Also, compounds that induce low-spin Soret band shifts are usually hydroxylation products of P450 catalysis, or potent P450 inhibitors.49Substrate concentration is a critical parameter in the design of the Soret shift assay. The Soret shift is largest at the substrate concentration where the enzyme is completely saturated by the substrate (but not over-saturated.50,51) However at saturating concentrations, the assay loses its usefulness to distinguish between substrate–enzyme pairs with more or less affinity for binding and causing transition to the catalytically active high-spin form. Furthermore, enzyme-saturating levels of substrate in free solution may not always be achievable due to solubility limitations, as was true for the benzocycloarene compounds used here. Therefore, we adjusted the substrate concentration to a single value for all benzocycloarene–enzyme pairs so that the Soret shift was readily measured, but not optimized for any single substrate–enzyme combination. This concentration, 50 μM, is well below the solubility limit of all the benzocycloarenes used here, but provided an easily measured Soret shift that could be interpreted as a relative measure of binding affinity and extent of high-spin shift.
Fig. 3 shows absorbance spectra for wild type P450cam without camphor, which is the reference state for the completely low-spin state, and with bound camphor, which is the reference state for the completely high-spin state. The high-spin and low-spin states exhibit the characteristic Soret absorption peaks at 417 and 391 nm, respectively. The spin state of unbound P450cam is taken to be 0% HS, and the spin state of wild type P450cam with 0.2 mM camphor is taken to be 100% HS.31 Since Y96F does not exhibit a complete shift to high spin upon binding camphor, the WT+camphor was also used to represent 100% HS for measurements involving the mutant Y96F P450cam. Also shown in Fig. 3 are the spectra for the BCA6-enzyme pairs: WT+tetralin and Y96F+tetralin. For each benzocycloarene-enzyme pair, the percent high spin (%HS)
was computed from:
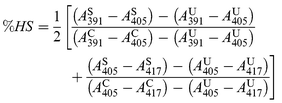 | (1) |
=(5±2)
%HS, BCA5=(10±2)
%HS, BCA6=(10±2)
%HS, BCA7=(18±2)
%HS] were found to be well below the values for Y96F
P450cam [BCA4=(31±1)
%HS, BCA5=(53±2)
%HS, BCA6=(58±2)
%HS, BCA7=(61±2)
%HS].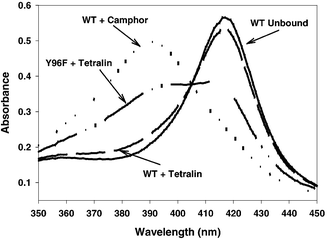 | ||
| Fig. 3 Absorbance spectra (5 μM protein) for WT P450cam with and without camphor (WT+camphor, WT Unbound, respectively); with BCA6 (WT+tetralin); and for mutant Y96F P450cam with BCA6 (Y96F+tetralin). The spectra with camphor (0.2 mM camphor) or without substrate are in buffer T. Spectra with BCA6 contain 50 μM BCA6 in 100 mM DMSO. | ||
Fig. 4 Compares the %HS values with the normalized NADH coupling efficiencies and maximum product turnover rates. The calculated %HS values show a clear correlation to both NADH coupling and product turnover. This result suggests that by using sub-saturating concentrations of potential substrates, a simple fast spectroscopic method might be developed for high-throughput screening of P450 biocatalysis targets.
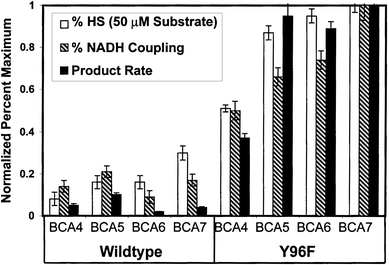 | ||
| Fig. 4 Comparison of normalized Soret shift assay values (%HS) with NADH coupling and product turnover rates for each P450cam-substrate pair. | ||
Conclusions
P450cam monooxygenation generally yields fewer products from each substrate than does the equivalent catalysis performed by either toluene or naphthalene dioxygenases. P450cam is the only demonstrated catalyst for oxygenating this class of substrates when the alkane ring size is 6 carbons or more. There are also differences in the regio- and stereospecificity of the products formed by each of the three enzymes. The observed regiospecificity for P450 products can be rationalized in terms of the relative radical stabilities of each potential hydroxylation site. Molecular dynamics simulations were in agreement with the above finding in that each substrate was mobile in the active site. NADH coupling efficiencies, binding affinities, and product turnover rates for the benzocycloarenes were found to be much higher for the Y96F mutant enzyme, indicating that the removal of the active site tyrosine hydroxyl resulted in an enzyme better suited for hydroxylation of hydrophobic compounds. In addition, NADH coupling, binding affinity, and product turnover rate were shown to increase with arene ring size in most cases. The insignificant level of correlation between product turnover and substrate binding indicated that binding alone was not sufficient to determine catalytic activity. A simple spectrophotometric assay that takes advantage of the Soret band shift induced by substrate binding qualitatively predicted the levels of catalysis for each benzocycloarene–enzyme pair investigated. This Soret shift assay could be developed into a useful tool for identifying potential targets of P450 catalysis in a high-throughput screening format.Acknowledgements
We thank Professor Derek Boyd for a gift of benzocyclohepten-3-ol.References
- H. L. Holland, in Stereoselective Biocatalysis, ed. R. N. Patel, Marcel Dekker, Inc., New York, NY, 2000, ch. 5..
- L. P. Wackett, L. D. Kwart and D. T. Gibson, Biochemistry, 1988, 27, 1360 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, T. A. Evans, M. Groocock, J. F. Malone, P. J. Stevenson and H. Dalton, J. Chem. Soc., Perkin Trans. 1, 1997, 1879..
- A. D. Grund, F. L. Hedberg and L.-S. Tan, in Plastics from Microbes: Microbial Synthesis of Polymers and Polymer Precursors ed. D. P. Mobley, Hanser/Gardner Publications, Inc, Cincinnati, OH, USA, 1994, ch. 6..
- D. T. Gibson, S. M. Resnick, K. Lee, J. M. Brand, D. S. Torok, L. P. Wackett, M. J. Schocken and B. E. Haigler, J. Bacteriol., 1995, 177, 2615 CAS.
- D. A. Grayson and V. L. Vilker, J. Mol. Catal. B: Enzym., 1999, 6, 533 CrossRef CAS.
- Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Plenum Press, New York, 1995..
- M. P. Mayhew, V. Reipa, M. J. Holden and V. L. Vilker, Biotechnol. Progr., 2000, 16, 610 CrossRef CAS.
- C. F. Oliver, S. Modi, M. J. Sutcliffe, W. U. Primrose, L. Y. Lian and G. C. K. Roberts, Biochemistry, 1997, 36, 1567 CrossRef CAS.
- K. Yanagita, I. Sagami and T. Shimizu, Arch. Biochem. Biophys., 1997, 346, 269 CrossRef CAS.
- V. V. Shumyantseva, T. V. Bulko, S. A. Alexandrova, N. N. Sokolov, R. D. Schmid, T. Bachmann and A. I. Archakov, Biochem. Biophys. Res. Commun., 1999, 263, 678 CrossRef CAS.
- D. P. Nickerson and L. L. Wong, Protein Eng., 1997, 10, 1357 CrossRef CAS.
- D. C. Lamb, D. E. Kelly, K. Venkateswarlu, N. J. Manning, H. F. J. Bligh, W.-H. Schunck and S. L. Kelly, Biochemistry, 1999, 38, 8733 CrossRef CAS.
- J. Cosme and E. F. Johnson, J. Biol. Chem., 2000, 275, 2545 CrossRef CAS.
- M. Shimoji, H. Yin, L. Higgins and J. P. Jones, Biochemistry, 1998, 37, 8848 CrossRef CAS.
- R. L. Wright, K. Harris, B. Solow, R. H. White and P. J. Kennelly, FEBS Lett., 1996, 384, 235 CrossRef CAS.
- L. S. Koo, R. A. Tschirret-Guth, W. E. Straub, P. Moenne-Loccoz, T. M. Loehr and P. R. Ortiz De Montellano, J. Biol. Chem., 2000, 275, 14112 CAS.
- J. K. Yano, L. S. Koo, D. J. Schuller, H. Li, P. R. Ortiz de Montellano and T.
L. Poulos, J. Biol. Chem., 2000, 275, 31086 CrossRef CAS.
- D. Harris and G. Loew, J. Am. Chem. Soc., 1995, 117, 2738 CrossRef CAS.
- J. J. De Voss and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 1995, 117, 4185 CrossRef CAS.
- J. A. Fruetel, J. R. Collins, D. L. Camper, G. H. Loew and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 1992, 114, 6987 CrossRef CAS.
- H. Lee, P. R. Ortiz de Montellano and A. E. McDermott, Biochemistry, 1999, 38, 10808 CAS.
- S. K. Ludemann, O. Carugo and R. C. Wade, J. Mol. Model., 1997, 3, 369 CrossRef CAS.
- M. D. Paulsen and R. L. Ornstein, Proteins, 1995, 21, 237 CAS.
- G. D. Szklarz and J. R. Halpert, Life Sci., 1997, 61, 2507 CrossRef CAS.
- P. J. Loida and S. G. Sligar, Biochemistry, 1993, 32, 11530 CAS.
- D. P. Nickerson, C. F. Harford-Cross, S. R. Fulcher and L. L. Wong, FEBS Lett., 1997, 405, 153 CrossRef CAS.
- J. A. Stevenson, J. K. Bearpark and L. L. Wong, New J. Chem., 1998, 22, 551 RSC.
- P. A. England, D. A. Rouch, A. C. G. Westlake, S. G. Bell, D. P. Nickerson, M. Webberley, S. L. Flitsch and L. L. Wong, Chem. Commun., 1996, 357 RSC.
- I. C. Gunsalus and G. C. Wagner, Methods Enzymol., 1978, 52, 166 Search PubMed.
- S. G. Sligar, Biochemistry, 1976, 15, 5399 CrossRef CAS.
- G. Wittig and W. Reuter, Liebigs. Ann. Chem., 1972, 765, 47 Search PubMed.
- D. A. Case, D. A. Pearlman, J. W. Caldwell, T. E. Cheatam III, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, D. M. Ferguson, R. J. Radner, G. L. Seibel, U. C. Singh, P. K. Weiner and P. A. Kollman, AMBER 6.0, University of California, San Francisco, CA, 1999..
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1996, 118, 2309 CrossRef CAS.
- J. R. Collins, D. L. Camper and G. H. Loew, J. Am. Chem. Soc., 1991, 113, 2736 CrossRef CAS.
- C. E. Schafmeister, W. S. Ross and V. Romanovski, Leap, University of California, San Francisco, CA, 1995..
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269 CAS.
- R. Raag and T. L. Poulos, Biochemistry, 1991, 30, 2674 CrossRef CAS.
- A. Roitberg and R. Elber, J. Chem. Phys., 1991, 95, 9277 CrossRef CAS.
- B. Das, V. Helms, V. Lounnas and R. C. Wade, J. Inorg. Biochem., 2000, 81, 121 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle, J. A. Pople, Gaussian 98, Gaussian, Inc., Pittsburgh, PA., 1998..
- D. A. Grayson, Y. B. Tewari, M. P. Mayhew, V. L. Vilker and R. N. Goldberg, Arch. Biochem. Biophys., 1996, 332, 239 CrossRef CAS.
- S. M. Resnick, K. Lee and D. T. Gibson, J. Ind. Microbiol., 1996, 17, 438 Search PubMed.
- M. Newcomb and P. H. Toy, Acc. Chem. Res., 2000, 33, 449 CrossRef CAS.
- B. Kauppi, K. Lee, E. Carredano, R. E. Parales, D. T. Gibson, H. Eklund and S. Ramaswamy, Structure, 1998, 6, 571 CrossRef CAS.
- R. E. White, M.-B. McCarthy, K. D. Egeberg and S. G. Sligar, Arch. Biochem. Biophys., 1984, 228, 493 CrossRef CAS.
- H. K. Anandatheerthavarada, S. Addya, J. Mullick and N. G. Avadhani, Biochemistry, 1998, 37, 1150 CrossRef CAS.
- M. R. Lefever and L. P. Wackett, Biochem. Biophys. Res. Commun., 1994, 201, 373 CrossRef CAS.
- S. Narasimhulu, Biochemistry, 1996, 35, 1840 CrossRef CAS.
- M. C. Marden and G. Hui Bon Hoa, Arch. Biochem. Biophys., 1987, 253, 100 CAS.
- S. Narasimhulu, L. M. Havran, P. H. Axelsen and J. D. Winkler, Arch. Biochem. Biophys., 1998, 353, 228 CrossRef CAS.
Footnotes |
| † Present address: Quantum Theory Project and Department of Chemistry, University of Florida, Gainesville, Florida. E-mail: Adrian@qtp.ufl.edu |
| ‡ Certain commercial equipment, instruments, and materials are identified in this paper to specify adequately the experimental procedure. In no case does such identification imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the material or equipment is necessarily the best available for the purpose. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2002 |