Design, synthesis, structural analysis and atropoisomerisation studies of polynucleating ligands based on porphyrins bearing catechol units†
Bertrand
Zimmer
,
Véronique
Bulach
,
Cathy
Drexler
,
Sabine
Erhardt
,
Mir Wais
Hosseini
* and
André
De Cian
Laboratoire de Chimie de Coordination Organique, Université Louis Pasteur, F-67000, Strasbourg, France. E-mail: hosseini@chimie.u-strasbg.fr
First published on 3rd January 2002
Abstract
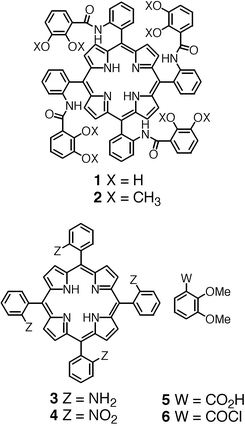
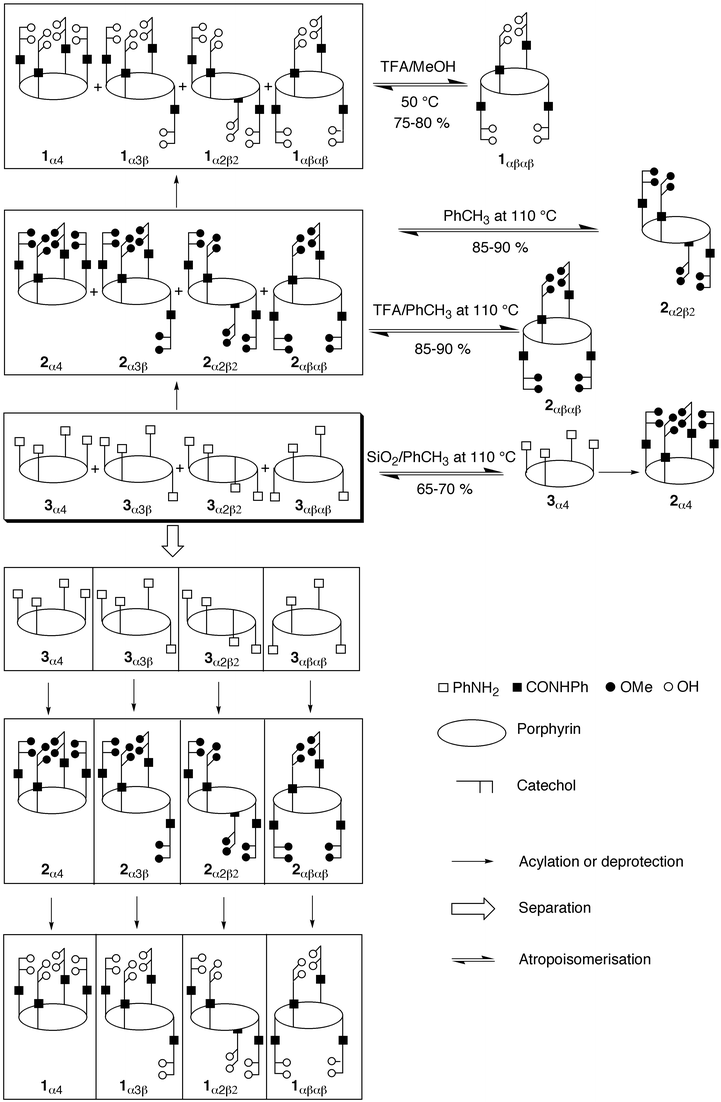
New polynucleating ligands based on a combination of a meso-tetrakis(phenyl)porphyrin core and four catecholamido units connected at the ortho positions of the phenyl moieties have been prepared. All four atropoisomers of the methoxy-protected meso-tetrakis(o-catecholamidophenyl)porphyrin 2 were obtained in high yields, however, the separation of isomers, although achievable, was found to be extremely tedious and did not allow the preparation of large quantities of the different atropoisomers. This difficulty was overcome by first separating the atropoisomers of the precursor meso-tetrakis(o-aminophenyl)porphyrin 3 and then by a quantitative condensation reaction of the separated isomers with the acyl chloride derivative of methyl-protected catechol 6. All four atropoisomers were subsequently deprotected in quantitative yields using BBr3. Although following
this route the protected 2α4 and 2α3β atropoisomers were obtained on the gram scale, for the other two 2αβαβ and 2α2β2 isomers, due to separation difficulties, only low quantities could be synthesised. After a systematic kinetic study of the atropoisomerisation process, the 2αβαβ isomer was obtained in large quantities by thermal isomerisation of the random mixture of isomers of 2 in toluene at 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C in the presence of a large excess of trifluoroacetic acid, whereas the 2α2β2 isomer was isolated under the same conditions but in the absence of acid. All four atropoisomers were characterised by classical 1- and 2-D NMR spectroscopy. Furthermore, the 1αβαβ, 1α4, 2αβαβ, 2α4
and 3αβαβ atropoisomers were also characterised in the solid state by X-ray diffraction on single crystals.
°C in the presence of a large excess of trifluoroacetic acid, whereas the 2α2β2 isomer was isolated under the same conditions but in the absence of acid. All four atropoisomers were characterised by classical 1- and 2-D NMR spectroscopy. Furthermore, the 1αβαβ, 1α4, 2αβαβ, 2α4
and 3αβαβ atropoisomers were also characterised in the solid state by X-ray diffraction on single crystals.
Porphyrins are among the most fascinating organic molecules. They have been employed in chemistry, biology, medicine and materials science.1 The porphyrin core binds a vast variety of metal cations and in some cases, these complexes show remarkable redox as well as photochemical properties. Furthermore, since the porphyrin skeleton may be functionalised at the β-pyrrolic and/or meso positions, a large variety of ligands and devices have already been designed and obtained. Considering the porphyrin core as a preorganised complexing unit, upon addition of further coordination sites, one may design a variety of di- and polynucleating ligands. For example, porphyrin derivatives bearing neutral bidentate units such as bipyridine and phenanthroline have been reported.2 On the other hand, porphyrins bearing dianionic bidentate catecholate units either directly connected at the meso positions3 or through a phenyl spacer unit have also been described.3,4 In the latter case, the porphyrin backbone was functionalised by 1 to 4 m-catecholamidophenyl units at the meso positions.4 Since the porphyrin core may be considered as a tetraazamacrocycle, one may draw an analogy with other systems combining polyaza-type ligands such as diaza-,5 triaza- and tetraaza-macrocycles,6 as well as a calix[4]arene derivative7 with the bidentate catechol unit.
In dealing with the design of new polynuclear complexes, in addition to the above-mentioned characteristics associated with the porphyrin unit, one may also take advantage of another particular feature, which is the possibility of obtaining stable atropoisomers by incorporating in the meso positions substituents that impose high rotational barriers.8 Atropoisomerisation has been elegantly exploited for the preparation of picket fence9 and other types of porphyrins.1 As previously reported,10 the meso-tetrakis(o-catecholamidophenyl)porphyrin 1 (Scheme 1) leads to four atropoisomers designed as 1α4, 1α3β, 1α2β2 and 1αβαβ (Scheme 2). The rationale behind the design of atropoisomers of 1 was based on the idea that whereas the 1α4 atropoisomer may lead to heterodinuclear complexes combining both transition and actinide or lanthanide metals in close proximity, the 1α2β2 and 1αβαβ isomers may form homo- or heterotrinuclear species with transition metals. Furthermore, for the 1αβαβ, 1α2β2 and 1α3β atropoisomers, the formation of infinite coordination polymers by simultaneous binding of transition and lanthanide metal cations accepting octa-coordination with cubic arrangements may be envisaged.
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
Here, we report a detailed kinetic study of the atropoisomerisation processes of derivatives of 1 and 2, detailed synthetic procedures, and structural studies on single crystals in the solid state.
Results and discussion
Design
The design of ligand 1 is based on a porphyrin core as a preorganised backbone and four catechol units as bidentate centres. The connection of four bidentate units to the porphyrin skeleton may be achieved at either β-pyrrolic or meso positions. Based on the distances between the catechol units, the meso position appeared to be the most suitable. For this choice, the catechol units may either be connected directly to the meso positions of the porphyrin or through spacer groups connected to the meso positions. For both geometrical and synthetic reasons, the phenyl group was chosen as the spacer. The attachment of the catechol units to the phenyl ring at the ortho positions with respect to the carbon atom connected to the porphyrin core was again based on the orientation of the catechol units and on the distances between them. Due to the stabilising effect towards oxidation of the amide group on the catechol, the connection of the bidentate units to the porphyrin core was achieved through amide junctions. Concerning the position of the amide group on the catechol, modelling suggested that the ortho position with respect to the hydroxyl group would be the most suitable, for orientational reasons. As mentioned above, owing to the bulkiness of the catecholamido group at the ortho position of the phenyl moiety, the four isolable atropoisomers 1α4, 1α3β, 1α2β2 and 1αβαβ may be expected.Synthetic strategy
Different synthetic strategies may be employed for the preparation of 1. One may synthesise the methyl-protected compound 2 by following the classical route based on an acid-catalysed condensation of pyrrole with a benzaldehyde derivative bearing at the ortho position the methyl-protected catecholamido group, followed by the deprotection the catechol units, leading thus to isomers of 1. Or alternatively, the synthesis of 1 may be based on the initial preparation of the previously reported meso-tetrakis(o-aminophenyl)porphyrin 3,9 then condensing the latter with the acyl chloride derivative of the methyl-protected catechol 611 leading to the protected isomers of 2 and finally deprotecting the catechol groups. It is worth noting that, since both strategies lead to the formation of 2 as a mixture of atropoisomers, upon deprotection of the catechol units a mixture of atropoisomers of 1 would be obtained. Thus, for both cases, the separation of atropoisomers appears crucial (Scheme 2).The synthesis of all four protected atropoisomers 2α4, 2α3β, 2α2β2 and 2αβαβ was first achieved by condensation of a statistical mixture of isomers of 3, obtained by the reduction of the nitro compound 4 prepared from ortho-nitrobenzaldehyde and pyrrole9, with 611 in THF and in the presence of Et3N. Although the condensation reaction proceeded quantitatively, separation of the mixture was found to be extremely tedious. Indeed, due to their low solubility, especially for the 2α2β2 isomer, and owing to the similar polarities of the 2α2β2 and 2αβαβ isomers, purification on a preparative silica column yielded, after at least three passages, only small quantities of the pure isomers. In an attempt to overcome this difficulty, the mixture of isomers was metallated by treating 2 with zinc acetate affording a mixture of zinc complexes (2-Zn). Again, the separation proved extremely difficult. Attempts to obtain better results using chromatotron or preparative HPLC were unsuccessful as well.
Confronted with the difficulty of preparing gram scale quantities of different atropoisomers of 2 and recognising that for atropoisomers of the precursor aminoporphyrin 3, especially the 3α2β2 and 3αβαβ isomers, the separation on the preparative scale was much easier, an alternative strategy based on the separation of all four atropoisomers of 39 and then condensation with the acyl chloride derivative of the methyl-protected catechol 6 was followed (Scheme 2). Although the acid-catalysed condensation of pyrrole with o-nitrobenzaldehyde, followed by reduction of the nitro groups to amino functionalities, was shown to afford under thermal equilibrium a statistical mixture (12.5, 50, 25, 12.5%) of the four atropoisomers 3α4, 3α3β, 3α2β2,
and 3αβαβ, respectively9, the ratio of different isomers may be tuned by external factors such as the solid support (SiO2) or the polarity of the solvent. For example, 3α4 may be obtained in 66% yield upon refluxing in the presence of silica gel the random mixture of atropoisomers in benzene.12 On the other hand, for the nitro precursor 4 of the ligand 3, the 4αβαβ isomer may be isolated in 72% yield by thermal equilibration of the random mixture of atropoisomers at 130°C in naphthalene; upon reduction of the nitro groups, 3αβαβ can be obtained in 39% yield.13 In a similar fashion, the preparation of 4α2β2 as its Zn complex has been described.14 Different atropoisomers of 3 were prepared according to the literature. In the case of 3αβαβ, since to the best of our knowledge no structural data is available, its solid state structure was also investigated by X-ray diffraction on a single crystal (see structural analysis below).
The condensation of the different pure isomers of 3 with 6 in the presence of Et3N was first achieved quantitatively at 25°C, however, at room temperature, isomerisation leading again to a mixture was observed. In order to minimise the atropoisomerisation, the condensation reaction was thus carried out at 0°C in THF and in the presence of Et3N. Again, the condensation reaction proceeded with almost quantitative yields. The pure atropoisomers 2αβαβ, 2α2β2, 2α3β and 2α4 could be obtained in 71, 61, 76 and 56% isolated yields, respectively. The deprotection of 2α4, 2α3β, 2α2β2 and 2αβαβ atropoisomers at −78°C using BBr3 in CH2Cl215
afforded the desired compounds 1α4, 1α3β, 1α2β2 and 1αβαβ in almost quantitative yields.
Following the above mentioned strategy, 2α4 and consequently 1α4 could be synthesized in gram quantity using Lindsey's method for the preparation of 3α4.12 On the other hand, since isomer 3α3β was present at 50% in the random mixture, isomer 1α3β could also be obtained in gram amounts. The other two isomers, 1αβαβ and 1α2β2, due to severe separation difficulties, were not directly accessible in large quantities. However, they could be prepared on the gram scale upon thermal atropoisomerisation (see below).
Structural and conformational analysis
| 1 α4 | 1 αβαβ | 2 α4 a | 2 αβαβ a | 3 αβαβ | |
|---|---|---|---|---|---|
| a Ref. 10. | |||||
| Chemical formula | 2(C72H50N8O12)·5CH2Cl2 | C72H50N8O12·4CH3OH | C80H66N8O12·2H2O | C80H666N8O12·2H2O | C44H34N8 |
| Formula weight | 2863.16 | 1345.40 | 1367.50 | 1570.2 | 674.82 |
| Crystal system | Monoclinic | Tetragonal | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c | I4/a | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2/1 |
| a/Å | 12.7436(2) | 22.2498(5) | 29.447(2) | 16.305(5) | 12.129(3) |
| b/Å | 12.9855(3) | 10.672(1) | 17.578(5) | 11.168(1) | |
| c/Å | 39.3596(9) | 13.3040(4) | 27.067(3) | 13.881(4) | 13.627(3) |
| α/deg | 105.29(2) | ||||
| β/deg | 92.368(5) | 123.449(6) | 100.09(2) | 108.60(2) | |
| γ/deg | 98.28(2) | ||||
| U/Å3 | 6507.7(4) | 6586.2(5) | 7096(2) | 3701.7 | 1749(1) |
| Z | 2 | 4 | 4 | 2 | 2 |
| μ(Mo-Kα)/mm−1 | 0.297 | 0.097 | 0.083 | 27.211 | 0.073 |
| T | 173 | 173 | 294 | 173 | 294 |
| Meas. reflect. | 15305 | 19284 | 7751 | 8276 | 3289 |
| Reflect with I>3σ(I) |
7282 | 3132 | 3111 | 6342 | 1293 |
| R | 0.071 | 0.062 | 0.087 | 0.063 | 0.043 |
| wR | 0.082 | 0.074 | 0.129 | 0.099 | 0.049 |
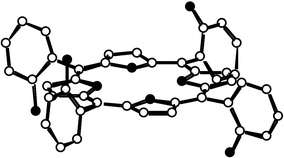 | ||
| Fig. 1 Crystal structure of the αβαβ atropoisomer of compound 3. H atoms are not shown for the sake of clarity. | ||
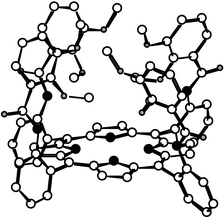 | ||
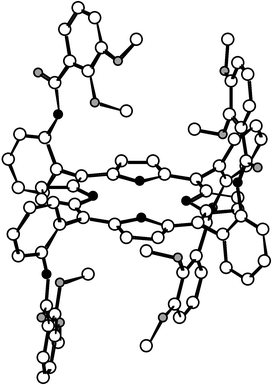
| Fig. 2 Crystal structure of the α4 atropoisomer of the catechol protected compound 2. Solvent molecules and H atoms are not shown for the sake of clarity. | ||
 | ||
| Fig. 3 Crystal structure of the 2αβαβ atropoisomer. Solvent molecules and H atoms are not shown for the sake of clarity. | ||
The X-ray analysis revealed the following common features for the solid state structures of compounds 2α4 and 2αβαβ: (i) as expected, in the case of 2α4 all four protected catechol units are localised on the same face of the molecule (Fig. 2) whereas for 2αβαβ the two sets of two protected catechols are localised below and above the mean plane of the porphyrin (Fig. 3); (ii) in both cases, the core of the porphyrin is almost planar, however, due to steric reasons, the deformation is slightly greater for 2α4; (iii) the amide groups [C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O distance ranging from 1.218 to 1.226 Å, N(CO)C angle of 118.0°, NCO angle ranging from 121.5 to 123.2° for 2α4; CO
distance 1.218 to 1.227 Å, N(CO)C and NCO angles ranging from 117.0 to 118.8 and 117.0 to 123.0°, respectively, for 2αβαβ] are in a trans configuration with the NH hydrogen atoms inwardly oriented towards oxygen atoms belonging to the protected catechol units and thus forming strong H bonds (N⋯O distance in the range 2.679–2.723 Å) and the CO groups are oriented in a divergent fashion towards the periphery of the porphyrin backbone; (iv) in both cases, the protected catechol units (dC–OMe between 1.369–1.453 Å for 2α4 and 1.377–1.439 Å for 2αβαβ) are found to be oriented in a convergent manner; (v) the phenyl ring of the catechol units is almost perpendicular with respect to the porphyrin core (NCCCmeso dihedral angle ranging
from 0.0° to −1.0° for 2α4 and 0.0° to −8.3° for 2αβαβ).
O distance ranging from 1.218 to 1.226 Å, N(CO)C angle of 118.0°, NCO angle ranging from 121.5 to 123.2° for 2α4; CO
distance 1.218 to 1.227 Å, N(CO)C and NCO angles ranging from 117.0 to 118.8 and 117.0 to 123.0°, respectively, for 2αβαβ] are in a trans configuration with the NH hydrogen atoms inwardly oriented towards oxygen atoms belonging to the protected catechol units and thus forming strong H bonds (N⋯O distance in the range 2.679–2.723 Å) and the CO groups are oriented in a divergent fashion towards the periphery of the porphyrin backbone; (iv) in both cases, the protected catechol units (dC–OMe between 1.369–1.453 Å for 2α4 and 1.377–1.439 Å for 2αβαβ) are found to be oriented in a convergent manner; (v) the phenyl ring of the catechol units is almost perpendicular with respect to the porphyrin core (NCCCmeso dihedral angle ranging
from 0.0° to −1.0° for 2α4 and 0.0° to −8.3° for 2αβαβ).
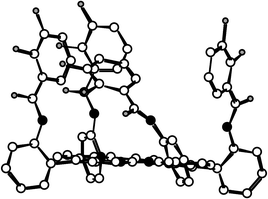 | ||
| Fig. 4 Crystal structure of the 1α4 atropoisomer. Solvent molecules and H atoms are not shown for the sake of clarity. | ||
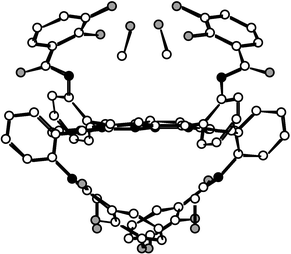 | ||
| Fig. 5 Crystal structure of the 1αβαβ atropoisomer. MeOH molecules bridging the catechol units through H bonds are represented. H atoms are not shown for the sake of clarity. | ||
The X-ray analysis revealed the following features for the solid state structure of 1α4: (i) one out of the four catechol units (dC–OH=1.342–1.372 Å) is disordered; (ii) as expected, all four catechol units are localised on the same face of the molecule (Fig. 4); (iii) the core of the porphyrin is almost planar; (iv) the amide groups [CO distance ranging from 1.128 to 1.230 Å, N(CO)C angle of 115.4–126.0°, NCO angle ranging from 121.5 to 123.2°] are in trans configuration with NH hydrogen atoms inwardly and the CO groups outwardly oriented. However, instead of NH⋯O hydrogen bonds as observed in the protected isomers of 2, OH⋯O hydrogen bonds are formed between the oxygen atom of the carbonyl group and the adjacent OH group of the catechol
moiety (O–O distance in the range 2.52–2.60 Å). This due owing to the rotation of all four catechol units, leading to an orientation of the OH groups towards the periphery of the porphyrin backbone.
The analysis in the solid state of the structure of the 1αβαβ atropoisomer revealed the following characteristics: (i) as expected from the NMR data, the two sets of two catechol units (dC–OH=1.368 Å) are localised below and above the mean plane of the porphyrin (Fig. 5); (ii) the core of the porphyrin presents some slight distortion with respect to planarity; (iii) the amide groups [dC–O=1.230 Å, N(CO)C and NCO angles of 115.5 and 123.0°, respectively] are in trans configuration with NH hydrogen atoms inwardly and the CO groups outwardly oriented; (iv) in contrast with the 1α4 atropoisomer mentioned above, the catechol units are found to be oriented in a convergent manner; (v)
the phenyl rings are tilted with respect to the mean plane of the porphyrin core (NCCCmeso dihedral angle of 6.0°); (vi) probably due to the presence of a hydrogen bond of the NH⋯OH type (dNH–O=2.640 Å) on the one hand and the presence of MeOH molecules, bridging two catechol units on each face of the porphyrin core through strong hydrogen bonds of the OH⋯O type (dO–O of 2.706 and 2.748 Å), the two catechols on the same side of the porphyrin backbone are facing each other.
UV-VIS absorption spectroscopy
All four 2α4, 2α3β, 2α2β2 and 2αβαβ isolated isomers showed almost identical absorption characteristics in CH2Cl2 at 25°C, that is a Soret band at ca. 420 nm and Q bands at 513, 545(sh), 588 and at 653 nm (Fig. 6). This observation is in agreement with the above mentioned solid state structure analyses indicating that the porphyrin cores are almost undeformed. However, for the diprotonated isolated atropoisomers 2-2H+, the Q band at 513 nm vanished, and significant red shifts of both the Soret and the other two Q bands were observed. The titration study was carried out by addition of increasing amounts of trifluoroacetic acid (TFA) to pure atropoisomers of 2 in CH2Cl2 at 25°C. For 2α4,
upon addition of TFA, whereas the Soret band appeared at 439 nm, the Q bands were present at 584 and 636 nm. For 2α3β, addition of TFA caused a 20 nm red shift of the Soret band, which appeared at 441 nm, whereas the Q bands were visible at 587 and 640 nm. For 2α2β2, in the presence of an excess of TFA, the Soret and Q bands appeared at 440, 586 and 639 nm, respectively. Finally, for the 2αβαβ isomer the addition of TFA caused the largest red shifts of both the Soret and Q bands, which appeared at 446, 598 and 651 nm, respectively. These rather large red shifts are indicative of out-of-plane deformation due to protonation of the porphyrin core, thus leading to a lower degree of conjugation of the π-system.16 This observation is in agreement with the previously reported solid state structure of a diprotonated porphyrin.17
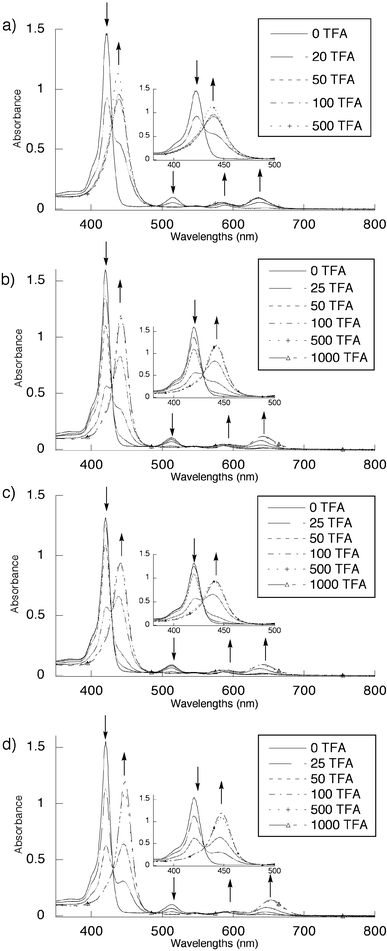 | ||
| Fig. 6 Protonation study of pure atropoisomers of 2 by UV-VIS absorption spectroscopy in CH2Cl2 at 25°C (see text). Arrows represent the increase and decrease of different absorption bands. (a)
2α4, (b)
2α3β, (c)
2α2β2 and (d)
2αβαβ. | ||
The titration plots (Fig. 7) clearly indicate that the basicity of the four different atropoisomers is rather different. Indeed, whereas diprotonation of the 2α4
[Fig. 7(a)] and 2α2β2
[Fig. 7(c)] isomers occurred upon addition of ca. 100 and 300 equiv. of TFA, respectively, for the other two isomers 2α3β
[Fig. 7(b)] and 2αβαβ
[Fig. 7(d)], the fully protonated species appeared upon addition of ca. 200 eq. of TFA. In other terms, half protonation occured after addition of ca. 21, 57, 64 and 71 equiv. of TFA for the 2α4, 2αβαβ, 2α3β
and 2α2β2, respectively, demonstrating the following order of basicity: 2α4>2αβαβ>2α3β>2α2β2.
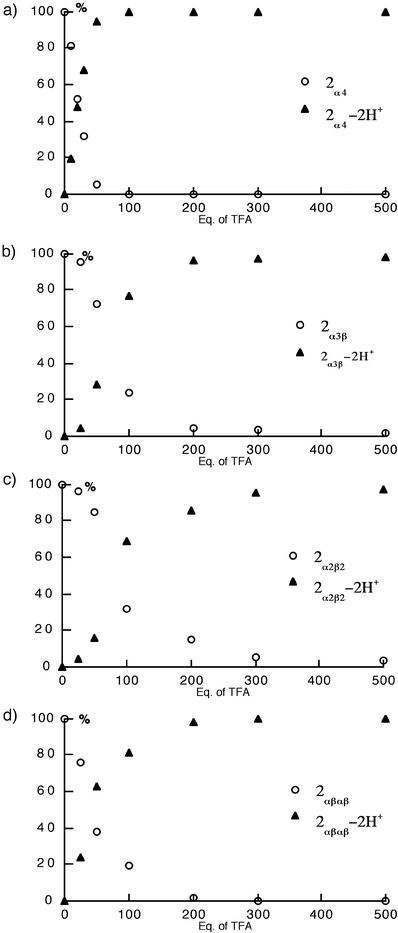 | ||
| Fig. 7 Trifluoroacetic acid (TFA) titration plots of pure atropoisomers of 2 in CH2Cl2 at 25°C. The percentage of the different species is based on the disappearance of the Soret bands for the unprotonated isomer 2
(○) and the appearance of the Soret bands for the 2-2H+ species (▲). (a)
2α4, (b)
2α3β, (c)
2α2β2 and (d)
2αβαβ. | ||
Atropoisomerisation
The rate of atropoisomerisation and the distribution of different isomers at equilibrium depend on the bulkiness of the substituents at the ortho position of the phenyl moiety, the deformability of the porphyrin core, the polarity of the medium, the presence of other chemicals and obviously the temperature. The role of the bulkiness of the substituents has been explained in terms of steric repulsion.18 Since the interconversion of different isomers requires the crossing of the substituents in the transition state,8 the deformation of the porphyrin backbone plays an important role in the isomerisation process. In particular, it has been demonstrated that upon double protonation, considerable deformation with respect to the planarity of the porphyrin core may be induced.16,17 It has been also shown that the proportion of the most polar and most sterically crowded α4 atropoisomer tends to decrease in non-polar solvents such as toluene, xylene or mesitylene, whereas the proportion of the least polar and the sterically least hindered αβαβ atropoisomer is increased.19 Furthermore, the distribution of different isomers may be altered by the presence of an external molecule capable of specific interactions with one of the isomers. This has been demonstrated in the case of meso-tetrakis-(2-hydroxy-4-nonylphenyl) porphyrin with tetramethoxy-p-benzoquinone, which favours (up to 78%) the formation of the α4 isomer.20
In the solid state, all separated atropoisomers of catechol protected 2 were found to be stable and no interconversion between them was observed. The same behaviour was also observed in solution in CHCl3 at room temperature and up to 50°C.
Study of a mixture of isomers of 2 with TFA
The atropoisomerisation was first studied on a statistical mixture of isomers of 2 (12.5, 50, 25, 12.5% of 2α4, 2α3β, 2α2β2, and 2αβαβ, respectively) obtained upon treatment of a statistical mixture of 3 by the acyl chloride derivative 6. The process was investigated in toluene at 110°C in the presence of trifluoroacetic acid (TFA). Due to its rather low polarity, toluene, in which all four isomers are soluble, appeared to be the best solvent since on the one hand it should favour the least polar and the sterically hindered 2αβαβ, and 2α2β2, atropoisomers, and on the other hand, it allows rather high temperatures to be used. Based on the above mentioned observations reported in the literature, the isomerisation process was
carried out on the mixture of protonated 2-2H+ species using TFA. The choice of the latter was based on its solubility in toluene. Owing to the difference in chemical shifts of the methoxy groups for different atropoisomers, the atropoisomerisation process was conveniently monitored by 1H-NMR and the proportion of the different isomers was determined by integration of the OCH3 signals (Fig. 8).
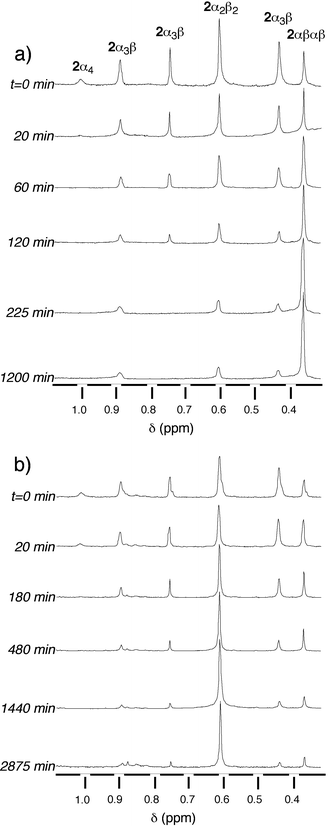 | ||
| Fig. 8 Time evolution of the atropoisomerisation of a statistical mixture of isomers of 2 in toluene at 110°C studied by 1H-NMR at (CDCl3, 25°C, 300 MHz). Only the region containing the signals corresponding to the methoxy groups is presented. (a) In the presence of 103 equiv. trifluoroacetic acid (TFA) and (b) in the absence of TFA. | ||
Starting with a statistical mixture of isomers, the effect of the concentration of TFA on the proportions of different atropoisomers of 2 was studied in toluene at 110°C (Fig. 9). After 4 days, the plot (% of different isomers vs. equiv. of TFA added) showed that both in the absence and presence of TFA, 2α4 entirely disappeared. At low acid concentration the major isomer was found to be 2α2β2, however, above a 60 equivalents excess of TFA, the 2αβαβ atropoisomer was the dominant species in the mixture. In the presence of a large excess of TFA (103 equiv.), the mixture reached after 4 days a plateau composed of ca. 90%
2αβαβ, 7%
2α3β and 3%
2α2β2
(Fig. 9).
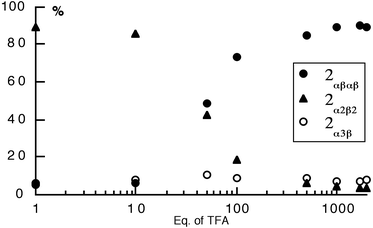 | ||
| Fig. 9 The effect of trifluoroacetic acid (TFA) concentration on the random mixture of atropoisomers of 2 in toluene at 110°C after 4 days. The proportion of different isomers was monitored by 1H-NMR in CDCl3. | ||
The kinetics of atropoisomerisation was studied in toluene at 110°C on the statistical mixture of isomers in the presence of 103 equiv. of TFA [Fig. 8(a)]. The plot (% of different isomers vs. time) shows that in the early stages of the process, the proportion of the 2α4, 2α3β and 2α2β2 isomers decreased rapidly, with a concomitant increase of the 2αβαβ atropoisomer. After ca. 10 h, the system reached an equilibrium and the mixture contained ca. 86%
2αβαβ, 7%
2α3β and 7%
2α2β2
[Fig. 10(a)].
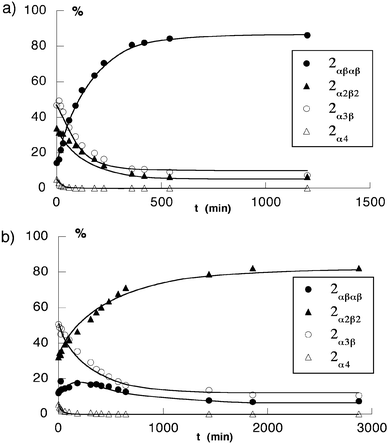 | ||
| Fig. 10 Kinetic plots of the atropoisomerisation of the statistical mixture of atropoisomers of 2 in toluene at 110°C. (a) in the presence of 103 equiv. trifluoroacetic acid (TFA) and (b) in the absence of TFA. The proportion of different isomers was monitored by 1H-NMR in CDCl3. | ||
°C, starting with statistical mixture of isomers, the plot (% of different isomers vs. time) indicates that in the early stages of the process, whereas the proportion of 2α4 and 2α3β decreased, the percentage of both the 2α2β2 and 2αβαβ isomers increased. After ca. 20 h, the system reached an equilibrium and the mixture contained ca. 80% of 2α2β2, 7% of 2αβαβ and 13% of 2α3β
[Fig. 10(b)]. So far, we are unable to propose a possible molecular explanation for this observation.
°C in toluene in the absence as well as in the presence of TFA (103 equiv.). Starting from the 2α4 isomer, in the absence of TFA, a rather rapid decrease in the percentage of 2α4 with concomitant build-up of the other three isomers was observed. The percentage of both the 2α3β and 2αβαβ isomers first increased to maximum values of ca. 70 and 30%, respectively, before decreasing, indicating that they are intermediates in the formation of the 2α2β2 isomer, which is the most abundant final species [Fig. 11(a)].
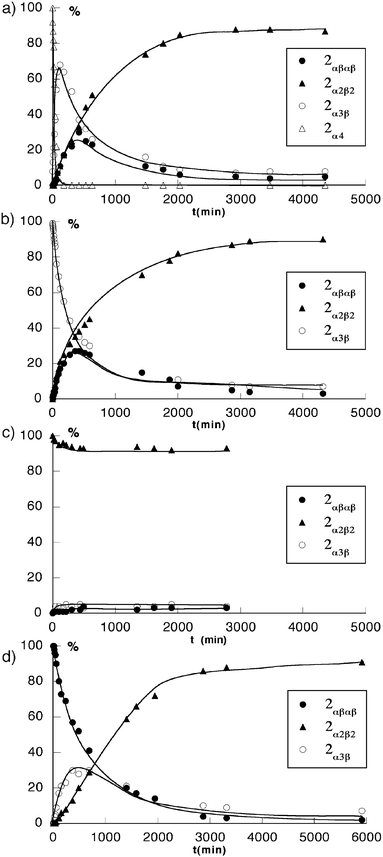 | ||
| Fig. 11 Kinetic plots of the atropoisomerisation of pure isomers of 2 under neutral conditions (toluene at 100°C), (a)
2α4, (b)
2α3β, (c)
2α2β2 and (d)
2αβαβ. The proportion of different isomers was monitored by 1H-NMR in CDCl3. | ||
When starting from the 2α3β isomer, the same type of behaviour was observed [Fig. 12(a)]. During the course of the isomerisation process, no 2α4 could be detected. The disappearance of the 2α3β isomer corresponded to the transient appearance of 2αβαβ, which reached a maximum of ca. 23% and finally, as in the previous case, the formation of 2α2β2 was observed [Fig. 11(b)].
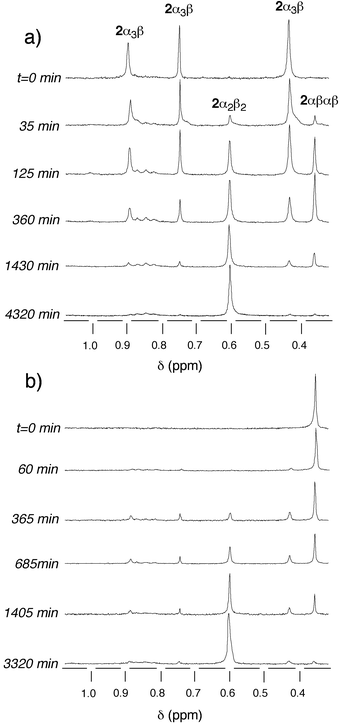 | ||
| Fig. 12 Time evolution of the atropoisomerisation of pure isomers of 2 in toluene at 110°C in the absence of acid studied by 1H-NMR at (CDCl3, 25°C, 300 MHz). Only the region containing the signals corresponding to the methoxy groups is presented. (a)
2α3β and (b)
2αβαβ. | ||
Similarly, when starting with the pure 2αβαβ isomer [Fig. 12(b)], whereas the latter disappeared, the 2α3β isomer appeared as a transient species (maximum of ca. 30%) and the most abundant final isomer was 2α2β2 [Fig. 11(d)].
Finally, the stability of the 2α2β2 isomer under the same conditions was established by heating it in toluene at 110°C for several days. In agreement with the above mentioned observations, a slight decrease in the percentage of 2α2β2 with the appearance of the other two 2α3β and 2αβαβ isomers in low percentages was observed [Fig. 11(c)].
°C.
Starting from the 2α4-2H+ isomer, again a rather fast decrease of the percentage of 2α4 and the formation of the other three isomers was observed [Fig. 13(a)]. However, in marked contrast with the study in the absence of TFA, the percentage of both 2α3β-2H+ and 2α2β2-2H+ isomers first increased to maximum values of ca. 68 and 30%, respectively and then the concentration of both isomers decreased, leading to the formation of 2αβαβ-2H+ as the dominant final species [Fig. 14(a)].
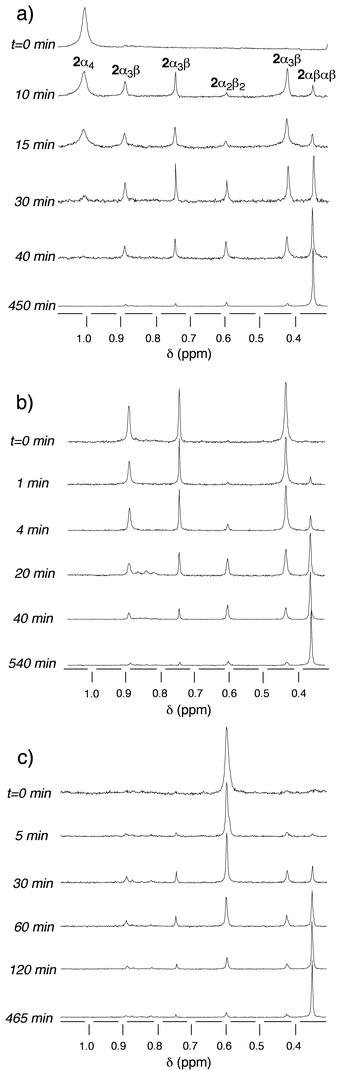 | ||
| Fig. 13 Time evolution of the atropoisomerisation of pure isomers of 2 in toluene at 110°C in the presence of 103 equiv. of TFA studied by 1H-NMR (CDCl3, 25°C, 300 MHz). Only the region containing the signals corresponding to the methoxy groups is presented. (a)
2α4−2H+, (b)
2α3β-2H+, (c)
2α2β2-2H+. | ||
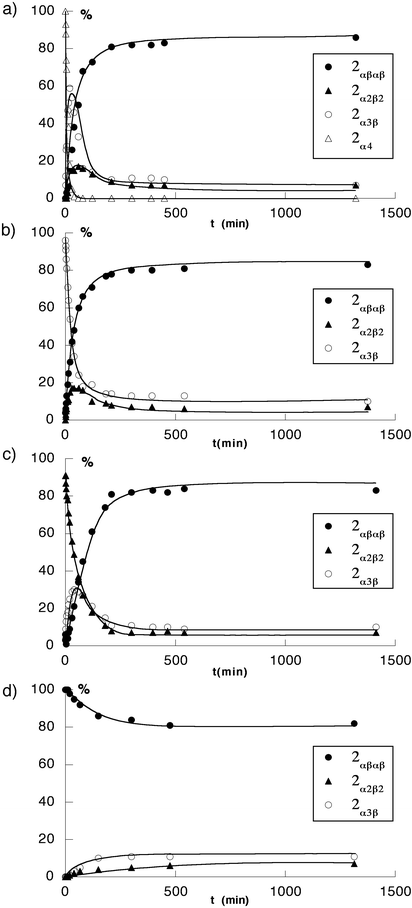 | ||
| Fig. 14 Kinetic plots of atropoisomerisation of pure isomers of 2 in the presence of 103 equiv. of TFA (toluene at 100°C). (a)
2α4-2H+, (b)
2α3β-2H+, (c)
2α2β2-2H+ and (d)
2αβαβ-2H+, The proportion of different isomers was monitored by 1H-NMR in CDCl3. | ||
When the starting species was the 2α3β isomer, the same type of behaviour was again observed [Fig. 13(b)]. No 2α4 isomer could be detected during the course of the isomerisation process. The disappearance of 2α3β-2H+, the common intermediate, corresponded to the appearance of both 2αβαβ-2H+ and 2α2β2-2H+. The latter, as in the previous case, was a transient species that reached a maximum of ca. 18% before decreasing, leading to the formation of 2αβαβ-2H+ as the dominant final isomer [Fig. 14(b)].
Similarly, when starting with pure 2α2β2-2H+ isomer [Fig. 13(c)], whereas the latter disappeared, the 2α3β isomer, the common intermediate, appeared as a transient species (maximum of ca. 34%) and the most abundant final isomer was as expected 2αβαβ-2H+
[Fig. 14(c)]. The stability of 2αβαβ-2H+ in the presence of TFA was demonstrated upon heating the pure 2αβαβ isomer in toluene at 110°C for several days. In agreement with the above mentioned observations, 2αβαβ-2H+ remained the dominant species although the appearance of the 2α3β-2H+
and 2α2β2-2H+ atropoisomers in low amounts was observed [Fig. 14(d)].
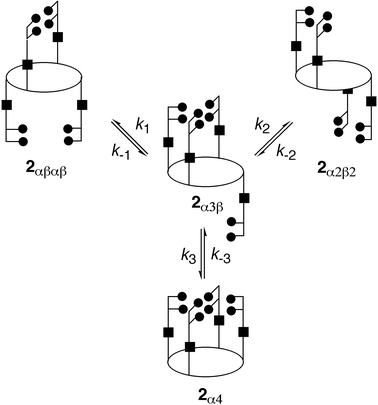 | ||
| Fig. 15 Kinetic scheme for the atropoisomerisation of 2 based on a single bond rotation model. 2α3β is considered to be the common intermediate. | ||
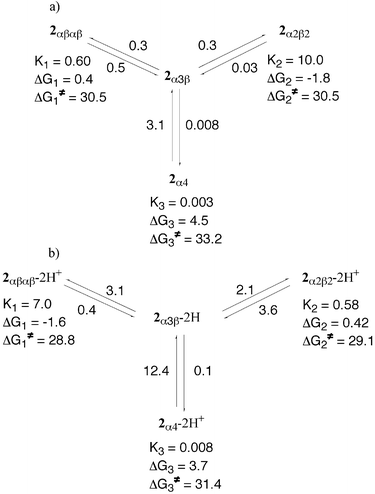 | ||
| Fig. 16 Schematic representation of the atropoisomerisation of 2 in toluene at 110°C in the absence (a) and presence (b) of TFA, summarising the calculated rate constants ki, equilibrium constants Ki, relative differences ΔGi and free energies of activation ΔGi≠. | ||
| No TFA | 103 equiv. TFA | |
|---|---|---|
|
a Values of average rate constants ki and k−i
(i=1–3) in 10−4 s−1
(see Fig. 15 for assignment of ki); values in parentheses are the standard deviation.
b Owing to the rather small number of data, values for k3 and k−3 are only indicative.
c Equilibrium constants Ki(i=1–3)=ki/k−i
(see Fig. 15 for assignment of Ki).
d Free energy of activation ΔGi≠
(i=1–3)
in kcal mol−1 calculated from the Eyring equation [ΔGi≠=−RTln(kih/kBT)].
e Relative free energy difference ΔGi
(i=1–3) in kcal mol−1 calculated using ΔGi=−RT lnKi
|
||
| k 1 a | 0.3 (0.1) | 3.1 (1.1) |
| k −1 a | 0.5 (0.1) | 0.4 (0.2) |
| k 2 a | 0.3 (0.05) | 2.1 (0.4) |
| k −2 a | 0.03 (0.01) | 3.6 (0.8) |
| k 3 a b | 0.008 | 0.1 |
| k −3 a b | 3.1 | 12.4 |
| K 1 c | 0.6 | 7.0 |
| ΔG1≠d | 30.5 | 28.8 |
| ΔG1e | 0.4 | −1.6 |
| K 2 c | 10.0 | 0.6 |
| ΔG2≠d | 30.5 | 29.1 |
| ΔG2e | −1.8 | 0.4 |
| K 3 c | 0.003 | 0.008 |
| ΔG3≠d | 33.2 | 31.4 |
| ΔG3e | 4.5 | 3.7 |
All six rate constants k1, k−1, k2, k−2, k3 and k−3
(see Fig. 15) could be calculated in toluene at 110°C. Whereas ki
(i=1–3) corresponds to the formation of the 2αβαβ, 2α2β2 and 2α4 atropoisomers, respectively, k−i(
i=1–3) represent their consumption. Although all four calculated rate constants for 2αβαβ and 2α2β2 are quite accurate, those for 2α4 are only indicative values because of its rapid disappearance at 110°C and thus the rather small
number of observed data. The range of observed rate constants is comparable with the results obtained previously for other types of porphyrins.16,22
In the absence of acid, the k1 and k−1 rate constants corresponding to the formation and disappearance of the 2αβαβ isomer are almost the same (ca. 0.4 0−4 s−1). On the other hand, for the 2α2β2 isomer, which is the final dominant species, the formation (k2) rate constant is almost identical to k1, whereas the consumption (k−2) one is an order of magnitude smaller, leading to a k2/k−2 ratio of ca. 10. Finally, in the case of 2α4, whereas k3 is smaller by a factor of ca. 40 than k1 and k2, k−3 is greater by almost an order of magnitude than k−1 or k−2 [Fig. 16(a)].
In the presence of 103 equiv. of TFA, as expected, the overall process was found to be faster by a factor of ca. 3. In marked contrast with the above mentioned observations in the absence of acid, the k1 and k−1 rate constants corresponding to the formation and disappearance of the 2αβαβ-2H+ isomer are different with a k1/k−1 ratio of ca. 10, whereas for the 2α2β2-2H+ isomer, k2 is lower than k−2 (k2/k−2 ratio of ca. 0.6). Again for 2α4-2H+, whereas k3 is smaller by a factor of ca. 30 and 20 than k1 and k2, respectively, the k3/k−3 ratio is calculated to be ca. 0.008 [Fig. 16(b)].
Both in the absence and in the presence of TFA, the equilibrium constants Ki
(i=1–3) could be calculated from the calculated rate constants ki using Ki=ki/k−i. In the absence of acid, the equilibrium constant K1 corresponding to the equilibrium between 2αβαβ and 2α3β is smaller by a factor of ca. 17 than K2 corresponding to the equilibrium between 2α2β2 and 2α3β, whereas K3 is considerably smaller than both K1 and K2. In the presence of TFA, for K1 and K2, the opposite trend is observed.
Indeed, K1 is greater than K2 by a factor of ca. 12, whereas for K3 the same trend is observed.
=−RT lnKi, the free energy difference for 2αβαβ, 2α2β2, 2α4 and for 2αβαβ-2H+, 2α2β2-2H+ and 2α4-2H+ with respect to 2α3β and 2α3β-2H+, respectively, was calculated in the absence and in the presence of TFA, respectively (Table 2). The range of values calculated (of the order of a few kcal mol−1) is comparable to previously reported results.22 Under neutral conditions, considering the 2α3β
isomer (the common intermediate) as the reference state, whereas 2αβαβ and 2α4 lie above it by ca. 0.4 and 4.5 kcal mol−1, respectively, the 2α2β2 isomer is found to be the most stable species (ΔG2 of ca.
−1.8 kcal mol−1). Under acidic conditions, whereas 2α4-2H+ remains the isomer with the highest free energy difference (ΔG3 of ca. 4 kcal mol−1), the reverse order is observed for the 2αβαβ-2H+ and 2α2β2-2H+ atropoisomers. Indeed, whereas a ΔG1 value of ca.
−1.6 kcal mol−1 was calculated for 2αβαβ-2H+,
a ΔG2 of ca. 0.4 kcal mol−1 was calculated for 2α2β2-2H+, indicating that the latter is less stable than 2αβαβ-2H+
[Fig. 16(b)].
Using the Eyring equation [ΔGi≠=−RT ln(kih/kBT)], the free energy of activation ΔGi≠ for 2αβαβ, 2α2β2, 2α4 and 2αβαβ-2H+, 2α2β2-2H+ and 2α4-2H+ was calculated in the absence and in the presence of TFA, respectively. The free energy of activation reflects the height of the rotational barrier. The calculated ΔGi≠ values range from ca. 29 to 33 kcal mol−1. These values are of the same order of magnitude as those previously reported for picket-fence
type porphyrins.16,22 When comparing 2 and 2-2H+ isomers, as expected, the ΔGi≠ values were higher under neutral conditions than in the presence of acid. Both in the absence and in the presence of TFA, ΔG1≠ and ΔG2≠, corresponding to a single rotation for 2αβαβ and 2α2β2
(ca. 30 kcal mol−1) or 2αβαβ-2H+ and 2α2β2-2H+
(ca. 29 kcal mol−1) are almost identical, indicating similar rotational barriers but lower than ΔG3≠ for 2α4 or 2α4-2H+.
For the deprotected isomers of 1, whereas in the solid state all four atropoisomers were found to be stable, in solution the protonated forms 1α4-2H+, 1αβαβ-2H+, 1α3β-2H+ and 1α2β2-2H+ were found to undergo atropoisomerisation. In deuterated MeOH at 50°C, after ca. 150 min, the 1α4-2H+ atropoisomer was found to be converted into a mixture composed of 74% of 1αβαβ-2H+, 19%
1α3β-2H+ and 7%
1α2β2-2H+ isomers (Fig. 17). Although this observation could have been interest for synthetic purposes, due to the presence of
unprotected catechol units, the separation of the three isomers present in the mixture was unsuccessful.
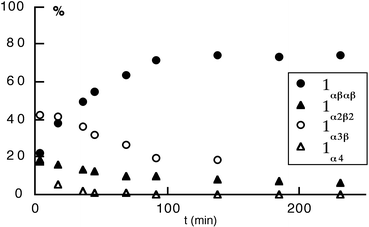 | ||
| Fig. 17 Kinetics of atropoisomerisation on a statistical mixture of isomers of 1 in CD3OD at 50°C: plot of the % of different isomers versus time. The proportion of different isomers was monitored by 1H-NMR (see text). | ||
Conclusions
In conclusion, four new polynucleating ligands based on a combination of a meso-tetrakis(phenyl)porphyrin core and four catecholamido units were prepared. Although all four atropoisomers of 2 were obtained in high yields, their separation was found to be extremely tedious and did not allow the preparation of large quantities of the different atropoisomers. This difficulty was overcome by first separating the atropoisomers of the precursor 3, followed by a quantitative condensation reaction of the separated isomers with 6. Although following that route the protected 2α4 and 2α3β atropoisomers could be obtained in gram quantities, for the other two 2αβαβ and 2α2β2 isomers, again due to separation difficulties, only low quantities could be synthesised.The isomerisation of all four atropoisomers was studied under thermal and acidic conditions. It was found that for the statistical mixture of protected isomers of 2, under neutral conditions, the atropoisomerisation in toluene at 110°C afforded 2α2β2 as the major species after 4 days. On the other hand, in the presence of TFA in excess, under otherwise identical conditions, the most abundant isomer was 2αβαβ-2H+. These observations were applied on a preparative scale, affording the 2αβαβ and 2α2β2 isomers in large quantities. All four atropoisomers were deprotected in quantitative yields using BBr3. All atropoisomers were characterised by classical 1- and 2-D NMR spectroscopy. Furthermore, in the case of the 1αβαβ, 1α4, 2αβαβ, 2α4
and 3αβαβ atropoisomers, their solid state structures were studied by X-ray diffraction on single crystals.
Using the prepared atropoisomers, the synthesis of discrete homo- and heteropolynuclear complexes with transition metals, lanthanides and actinides, as well as the formation of infinite coordination networks, are under current investigation.
Experimental
General details
When commercially available, all reagents were purchased and used without further purification. Reaction solvents were distilled prior to use using standard methods. Compounds were purified by column chromatography using Kiessel gel 60 (Merck; 43–60 mesh). 1H- and 13C-NMR spectra were recorded using Bruker spectrometers at 500, 400, 300 and 200 MHz and 50 MHz, respectively. 2-D ROESY spectra were recorded on a Bruker AMX 400 spectrometer.Microanalyses were performed by the “Service de Microanalyses de la Fédération de Recherche Chimie”, Université Louis Pasteur Strasbourg.
Syntheses
×75 ml), H2O (3×75
ml) and finally with 10% aqueous NaOH (3×75 ml) before the organic layer was dried over Na2SO4. After filtration and removal of the solvent, the solid was passed over an alumina column (Al2O3; CH2Cl2–MeOH 20 : 1) in order to remove excess acyl chloride. The intensely coloured reddish fractions containing the mixture of atropoisomers of 2 were collected and evaporated to dryness, affording a purple solid residue that was washed with n-hexane (75 ml), Et2O (75 ml) and dried under vacuum to give 3.3 g (84%) of the mixture of atropoisomers (SiO2; PhCH3–AcOEt 6 : 4, Rf=0.17 for 2α4, 0.47 for 2α3β, 0.60 for 2α2β2, 0.66 for 2αβαβ).
The isolation of pure atropoisomers using column chromatography (SiO2) proved to be very tedious due to solubility problems and small differences in the Rf values for the 2α2β2 and 2αβαβ atropoisomers and could only be achieved after several passages. The first passage over a silica column using a CCl4–CHCl3–AcOEt 5 : 1 : 1 mixture as the eluent afforded two fractions, one enriched in 2α4 and 2α3β atropoisomers and the other enriched in 2α2β2 and 2αβαβ atropoisomers. Upon a further two passages of the first fraction over a silica column using the same mixture as the eluent, pure 2α4
(10 mg) and 2α3β
(200 mg) atropoisomers were obtained in
0.2 and 5% yields, respectively. Another passage of the second fraction over a silica column (CCl4–CHCl3–AcOEt 5 : 2 : 1) afforded the pure 2α2β2
(15 mg) and 2αβαβ
(110 mg) atropoisomers in 0.4 and 2.8% yields, respectively.
2 αβαβ through condensation of 3αβαβ with 6. To a solution of 3αβαβ (100 mg, 0.15 mmol) and Et3N (170 µl, 1.22 mmol) in dry THF (50 ml), a large excess of 6 (370 mg, 1.84 mmol) was added at 0
°C and the mixture was stirred overnight at room temperature. After removal of the solvent, the solid residue was dissolved in CHCl3
(50 ml) and washed with 10% aqueous HCl (3×40 ml), H2O (3×40 ml), and finally with 10% aqueous NaOH (3×40 ml). The organic layer was dried over Na2SO4. After filtration
and evaporation of the solvent, column chromatography (SiO2; CH2Cl2–Et2O 93 : 7) of the solid residue afforded pure 2αβαβ
(140 mg) in 71% yield as a purple solid, which was further washed with n-hexane (50 ml), diethyl ether (50 ml) and finally dried under vacuum. 1H-NMR (CDCl3, 300 MHz, 25°C): δ
−2.49 (s, 2H, NH pyrrole), 0.37 (s, 12H, –OMe1), 2.87 (s, 12H, –OMe2), 6.51 (dd, 4H, J=1.5 and 8.1 Hz, Hcatechol), 6.85 (t, 4H, J=8.1 Hz, Hcatechol), 7.44 (ddd, 4H, J=1.1, 7.7 and 7.7 Hz, Hphenyl), 7.63 (dd, 4H, J=1.5 and 8.1 Hz, Hcatechol),
7.83 (m, 8H, Hphenyl), 8.85 (s, 8H, Hβ-pyrrole), 8.92 (dd, 4H, J=8.1 and 1.1 Hz, Hphenyl), 9.88 (s, 4H, NHCO); 13C-NMR (CDCl3, 75 MHz, 25°C): δ 55.22 (OMe), 58.11 (OMe), 115.79, 115.89, 122.11, 122.78, 123.25, 123.70, 125.88, 129.72, 131.70, 135.02, 139.35, 146.86, 151.40, 163.14 (NHCO); UV-VIS (CH2Cl2, 25°C): λmax/nm (ε/103) 374 sh (27), 420 (240), 512 (21), 545 sh (9.7), 587 (9), 651 (3.9); MS (FAB+): 1330.9 [M+]
(1330.5 calcd for C80H66N8O12); Anal. calcd for C80H66N8O12: C 72.17, H 5.00, N 8.42; found:
C 72.23, H 5.01, N 8.25%.
2 α2β2 through condensation of 3α2β2 with 6. The same procedure as for the preparation of 2αβαβ was followed and afforded 120 mg (61%) of the pure compound 2α2β2 after purification by column chromatography (SiO2; CH2Cl2, CH2Cl2–Et2O 93 : 7). 1H-NMR (CDCl3, 300 MHz, 25
°C): δ
−2.51 (s, 2H, NH pyrrole), 0.61 (s, 12H, –OMe1), 2.85 (s, 12H, –OMe2), 6.42 (dd, 4H, J=1.5 and 8.1 Hz, Hcatechol), 6.77 (t, 4H, J=8.1 Hz, Hcatechol), 7.43 (t, 4H, J=7.7
Hz, Hphenyl), 7.55 (dd, 4H, J=1.5 and 8.1 Hz, Hcatechol), 7.84 (m, 8H, Hphenyl), 8.85 (s, 4H, Hβ-pyrrole), 8.88 (s, 4H, Hβ-pyrrole), 8.96 (d, 4H, J=8.1 Hz, Hphenyl), 9.93 (s, 4H, NHCO); 13C-NMR (CDCl3, 75 MHz, 25°C): δ 55.17 (OMe), 58.66 (OMe), 115.91, 122.01, 122.71, 123.17, 123.62, 129.75, 131.65, 135.20, 139.20, 146.90, 151.40, 163.16 (NHCO); UV-VIS (CH2Cl2, 25°C): λmax/nm (ε/103)=374 sh (32), 420 (190), 512 (28), 545 sh (18), 584 (14), 652 (7.5).
2 α3β through condensation of 3α3β with 6. The same procedure as for the preparation of 2αβαβ was followed and afforded 150 mg (76%) of the pure compound 2α3β after purification by column chromatography (SiO2; CH2Cl2, CH2Cl2–Et2O 93 : 7 and finally CH2Cl2–Et2O 90 : 10). 1H-NMR (CDCl3, 300 MHz, 25
°C): δ
−2.50 (s, 2H, NH pyrrole), 0.44 (s, 6H, –OMe), 0.89 (s, 3H, –OMe), 0.93 (s, 3H, –OMe), 2.54 (s, 3H, –OMe), 2.88 (s, 6H, –OMe), 3.04 (s, 3H, –OMe), 6.28 (dd, 1H, J=1.5
and 8.1 Hz, Hcatechol), 6.50 (m, 3H, Hcatechol), 6.72 (t, 1H, J=8.1 Hz, Hcatechol), 6.83 (m, 3H, Hcatechol), 7.44 (m, 5H, 1Hcatechol and 4 Hphenyl), 7.61 (m, 3H, Hcatechol), 7.73 (dd, 1H, J=1.5 and 7.7 Hz, Hphenyl), 7.83 (m, 7H, Hphenyl); 8.91 (m, 12H, Hβ-pyrrole and 4 Hphenyl), 9.89 (s, 1H, NHCO), 10.06 (s, 1H, NHCO), 10.10 (s, 2H, NHCO); 13C-NMR (CDCl3, 75 MHz, 25°C): δ 54.43, 55.11, 55.32, 55.38, 58.39, 58.48, 58.82, 115.19, 115.27, 115.24, 115.51, 115.79, 115.87, 116.21, 116.34, 121.87, 122.05, 122.57, 122.73, 123.38, 123.59, 123.64, 125.65, 125.86, 125.94, 129.59, 131.52, 131.60, 131.67, 135.02, 135.23, 135.57, 138.72, 139.06,
146.44, 146.49, 146.60, 147.07, 147.15, 151.27, 151.32, 151.40, 162.90, 163.01, 163.24 (NHCO); UV-VIS (CH2Cl2, 25°C): λmax/nm (ε/103) 374 sh (27), 420 (240), 513 (21), 545 sh (9.6), 588 (9), 653 (4.2); MS (FAB+): 1330.9 [M+]
(1330.5 calcd for C80H66N8O12).
2 α4 through condensation of 3α4 with 6. The same procedure as for the preparation of 2αβαβ was followed and afforded 110 mg (56%) of the pure compound 2α4 after purification by column chromatography (SiO2; CH2Cl2, CH2Cl2–Et2O 80 : 20). 1H-NMR (CDCl3, 300 MHz, 25
°C): δ
−2.53 (s, 2H, NH pyrrole), 0.99 (s, 12H, –OMe1), 2.66 (s, 12H, –OMe2), 6.40 (d, 4H, J=8.1 Hz, Hcatechol), 6.80 (t, 4H, J=8.1 Hz, Hcatechol), 7.40 (dd, 4H, J=7.7 and 7.4 Hz,
Hphenyl), 7.63 (m, 8H, 4 Hphenyl and 4 Hcatechol), 7.83 (dd, 4H, J=7.7 and 7.0 Hz, Hphenyl), 8.89 (m, 12H, Hβ-pyrrole and Hphenyl), 10.31 (s, 4H, NHCO); 13C-NMR (CDCl3, 75 MHz, 25°C): δ 54.91 (OMe), 59.00 (OMe), 115.69, 116.19, 122.36, 122.96, 123.36, 123.70, 126.15, 129.62, 132.02, 135.96, 139.20, 147.07, 151.53, 163.22 (NHCO); UV-VIS (CH2Cl2, 25°C): λmax/nm (ε/103) 374 sh (24), 421 (220), 513 (20), 545 sh (9.5), 587 (8.7), 650 (3.8).
Compound 1αβαβ. To a solution of 2αβαβ (0.20 g, 0.15 mmol) in dry CH2Cl2 (50 ml) was added a 1 M solution of BBr3 in CH2Cl2 (7.5 ml, 7.5 mmol, 50 equiv.) at −78
°C. The mixture was gently allowed to reach room temperature and further stirred overnight. The mixture was again cooled to −78°C and MeOH (50 ml) was added to neutralise excess BBr3. The solution was stirred for another 4 h before the solvent was removed under reduced pressure. Excess BBr3 was co-evaporated with MeOH (at least 10×50 ml). The resulting solid was redissolved in MeOH (50 ml) and water (250
ml) was added. The mixture was extracted several times with CHCl3 until a colourless aqueous layer was obtained. The different collected organic layers were mixed, washed with H2O (3×50 ml), dried over MgSO4, filtered and evaporated to dryness. The residual solid was further washed with hexane (100 ml) before it was dried under vacuum overnight, affording the desired compound 1αβαβ
(0.11 g) as a purple solid in 60% yield. 1H-NMR (CD3OD, 500 MHz, 25°C): δ 6.13 (dd, 4H, J=8.1 and 7.7 Hz, Hcatechol), 6.38 (dd, 4H, J=7.7 and 1.5 Hz, Hcatechol), 6.66 (dd, 4H, J=8.1 and 1.5 Hz, Hcatechol), 7.49 (ddd, 4H, J=7.6, 7.6
and 1.1 Hz, Hphenyl), 7.79 (ddd, 4H, J=8.5, 7.6 and 1.4 Hz, Hphenyl), 7.84 (dd, 4H, J=7.6 and 1.4 Hz, Hphenyl); 8.53 (dd, 4H, J=8.5 and 1.1 Hz, Hphenyl), 8.80 (br s, 8H, Hβ-pyrrole); due to fast H/D exchange, pyrrolic NH, –OH and NHCO protons are not observed; 13C-NMR (CD3OD, 125 MHz, 25°C): δ 117.2, 118.4, 119.2, 119.6, 120.4, 124.9, 125.2, 130.5, 135.2, 137.0, 140.0, 146.1, 147.1, 167.9 (NHCO); UV-VIS (CH3OH, 25°C): λmax/nm (ε/103) 421 (210), 518 (20), 549 sh (11), 588 (9), 650 (4.7).
Compound 1α2β2. 2 α2β2 (70.0 mg, 0.053 mmol) was deprotected into 1α2β2 (0.032 g) in 50% yield following the procedure described above for the conversion of 2αβαβ into 1αβαβ. 1H-NMR (CD3OD, 500 MHz, 25
°C): δ 5.93 (dd, 4H, J=8.1 and 7.6 Hz, Hcatechol), 6.23 (dd, 4H, J=7.6 and 1.5 Hz, Hcatechol), 6.47 (dd, 4H, J=8.1 and 1.5 Hz, Hcatechol), 7.58 (ddd, 4H, J=8.1, 7.8 and 1.1 Hz, Hphenyl), 7.86 (ddd, 4H, J=8.1, 8.1 and 1.5 Hz, Hphenyl),
7.95 (dd, 4H, J=7.8 and 1.5 Hz, Hphenyl); 8.53 (dd, 4H, J=8.1 and 1.1 Hz, Hphenyl), 8.86 (br s, 8H, Hβ-pyrrole); due to fast H/D exchange, pyrrolic NH, –OH and NHCO protons are not observed; 13C-NMR (CD3OD, 125 MHz, 25°C): δ 118.0, 119.1, 119.5, 119.9, 124.9, 125.2, 130.7, 135.2, 137.0, 140.0, 146.1, 147.3, 168.2 (NHCO); UV-VIS (CH3OH, 25°C): λmax/nm (ε/103) 421 (190), 516 (17), 547 sh (7.6), 586 (7), 651 (3.1).
Compound 1α3β. 2 α3β (105 mg, 0.079 mmol) was transformed into 1α3β (0.040 g) in 42% yield following the procedure described above for the conversion of 2αβαβ into 1αβαβ. 1H-NMR (CD3OD, 300 MHz, 25
°C): δ 5.78 (dd, 1H, J=8.3 and 7.9 Hz, Hcatechol), 5.86 (dd, 2H, J=8.3 and 7.9 Hz, Hcatechol), 6.13 (m, 3H, Hcatechol), 6.34 (m, 5H, Hcatechol), 6.72 (dd, 1H, J=8.3 and 1.5 Hz, Hcatechol), 7.52 (m, 4H, Hphenyl), 7.81 (m, 6H, 2 Hphenyl), 7.92 (m, 2H, Hphenyl),
8.41 (dd, 2H, J=8.5 and 1.2 Hz, Hphenyl), 8.54 (dd, 1H, J=8.5 and 1.2 Hz, Hphenyl), 8.61 (dd, 1H, J=8.5 and 1.2 Hz, Hphenyl), 8.82 (br s, 8H, Hβ-pyrrole); 13C-NMR (CD3OD, 125 MHz, 25°C): δ 116.9, 117.3, 117.4, 117.5, 117.7, 118.6, 119.1, 119.2, 119.3, 199.4, 119.5, 119.6, 120.7, 124.4, 124.6, 124.9, 125.0, 125.5, 125.6, 130.5, 130.6, 130.7, 134.7, 135.1, 136.7, 137.0, 137.2, 139.7, 140.0, 140.1, 146.0, 146.2, 146.8, 147.4, 148.0, 167.6 (NHCO), 168.3 (NHCO), 168.8 (NHCO); UV-VIS (CH3OH, 25°C): λmax nm (ε/103) 422 (173), 516 (13.5), 548 sh (6.9), 587 (5.9), 651 (2.8).
Compound 1α4. 2 α4 (1.00 g, 0.75 mmol) was converted into 1α4 (0.84 g) in 92% yield following the procedure described above for the conversion of 2αβαβ into 1αβαβ. 1H-NMR (CD3OD, 500 MHz, 25
°C): δ 5.64 (dd, 4H, J=8.3 and 7.8 Hz, Hcatechol), 6.08 (dd, 4H, J=8.3 and 1.4 Hz, Hcatechol), 6.18 (dd, 4H, J=8.1 and 1.4 Hz, Hcatechol), 7.55 (ddd, 4H, J=7.6, 7.4 and 1.1 Hz, Hphenyl), 7.84 (m, 8H, Hphenyl), 8.45 (dd, 4H, J=8.6 and 1.1 Hz, Hphenyl),
8.89 (br s, 8H, Hβ-pyrrole); due to fast H/D exchange, pyrrolic NH, –OH and NHCO protons are not observed; 13C-NMR (CD3OD, 125 MHz): δ 117.1, 117.4, 118.8, 119.4, 119.5, 125.5, 129.9, 130.7, 136.0, 137.2, 139.4, 146.3, 148.4, 169.3 (NHCO); UV-VIS (CH3OH, 25°C): λmax/nm (ε/103) 421 (180), 515 (14), 548 sh (6.8), 588 (6.1), 649 (2.8); Anal. calcd for C72H50N8O12·CH2Cl2·CH3OH: C 66.51, H 4.23, N 8.39, O 15.57; found: C 66.85, H 4.35, N 8.03, O 15.67%.
Atropoisomerisation for synthetic purposes
×75 ml) and H2O (3×75 ml) before it was dried over MgSO4. After filtration and evaporation of the solvent, the residue was analysed by 1H NMR spectroscopy, which revealed the presence of 2αβαβ
(90%), 2α3β
(7%) and 2α2β2
(3%). After a first passage over an
SiO2 column (CH2Cl2 and CH2Cl2–Et2O 93 : 7), two sets of red fractions were collected, which after evaporation to dryness were washed with n-hexane (50 ml) and dried under vacuum. Whereas the first fraction (0.21 g) contained the pure 2αβαβ atropoisomer, the second one (0.08 g) was a mixture of 69%
2αβαβ, 8%
2α2β2 and 23%
2α3β. After further separation of the mixture on SiO2, 0.04 g of the desired 2αβαβ atropoisomer was obtained, thus increasing the yield to 83%
(0.25 g).
°C for 4 days. The solvent was evaporated and the crude solid was analysed by 1H-NMR spectroscopy, which revealed the presence of 2α2β2, 2α3β and 2αβαβ atropoisomers in 89%, 6.5% and 4.5% yields, respectively. Interestingly, due to the very low solubility of 2α2β2 in Et2O, when the solid containing the above mentioned mixture was washed with Et2O, both 2αβαβ and 2α3β isomers were eliminated, leading thus to the pure 2α2β2
(0.51
g) isomer in 85% yield.
Atropoisomerisation kinetic studies
For compound 2, the kinetics of the isomerisation process were studied on both the statistical mixture of isomers of 2 and on pure atropoisomers in toluene at 110°C, either in the presence or absence of trifluoroacetic acid. For the acid-catalysed process, typically 0.20 g of either the statistical mixture of 2 or pure isomers were dissolved in toluene (35 ml), 103 equiv. TFA were added and the solution heated in an oil bath. When the temperature reached 110°C, aliquots (∼1.5 ml) were removed at various times. The samples were evaporated to dryness, redissolved in CH2Cl2
(3 ml) and washed with 10% aqueous sodium hydroxide (10 ml). The organic layer was again evaporated to dryness. The percentage of different isomers was determined by 1H-NMR measurements by integration of the OMe proton signals in CDCl3: 0.99 ppm (s, 12H)
for 2α4, 0.44 (s, 6H), 0.89 (s, 3H) and 0.93 ppm (s, 3H) for 2α3β, 0.61 ppm (s, 12H) for 2α2β2, 0.37 ppm (s, 12H) for 2αβαβ.
In the absence of TFA, a solution of 0.20 g of either the statistical mixture of isomers of 2 or pure isomers in toluene (35 ml) was heated with an oil bath to 110°C. When temperature equilibration was reached, aliquots (∼1.5 ml) were removed at different time intervals and evaporated to dryness. The percentage of different isomers was, as indicated above, monitored by 1H-NMR in CDCl3.
The effect of the concentration of TFA on the distribution of different isomers of 2 was studied on the statistical mixture in toluene at 110°C. A solution of 0.1 g of the mixture and x equiv. of TFA (x=10, 50, 100, 500, 1000, 1700, 2000) in toluene (25 ml) was heated to 110°C in an oil bath for 4 days. The mixture was then evaporated to dryness and the residue dissolved in CH2Cl2
(40 ml) and washed with 10% aqueous sodium hydroxide (20 ml) and water (2×20 ml) before it was dried over MgSO4. After evaporation to dryness, the solid was dissolved in CDCl3 and the composition of the mixture was monitored by 1H-NMR using the integration of the OMe proton signals.
For the statistical mixture of isomers of 1 used as their hydrobromide salts, the isomerisation process was carried out in deuterated methanol at 50°C on a 4 h time scale and was monitored by 1H-NMR spectroscopy. The percentage of the different isomers was determined by integration of the β-pyrrolic proton signals (8.66 ppm (s, 8H) for 1α4·2HBr, 8.90 (m, 4H), 8.54 (d, 2H) and 8.79 ppm (d, 2H) for 1α3β·2HBr, 8.94 (s, 4H) and 8.93 ppm (s, 4H) for 1α2β2·2HBr, 8.98 ppm (s, 8H) for 1αβαβ·2HBr.
X-Ray crystal structure characterisation
For the protected 2α4 and 2αβαβ atropoisomers, the X-ray studies have been reported previously in a communication (CCDC reference number 182/775).10Suitable single crystals for the 3αβαβ atropoisomer were obtained upon slow liquid–liquid diffusion of MeOH into a chloroform solution containing 3αβαβ. The data was collected at room temperature on a Nonius MACH 3 diffractometer using Mo-Kα graphite-monochromated radiation.
For crystals of the 1α4 and 1αβαβ isomers, X-ray diffraction data collection was carried out at 173 K on a Kappa CCD diffractometer, equipped with an Oxford Cryosystem liquid N2 device, using graphite-monochromated Mo-Kα radiation.
For all structures, diffraction data were corrected for absorption and analysed using the OpenMolen package.23 All non-H atoms were refined anisotropically. Crystal data and details of measurements are reported in Table 1.
CCDC reference numbers 173460–173462. See http://www.rsc.org/suppdata/nj/b1/b104084g/ for crystallographic data in CIF or other electronic format.
Acknowledgements
G. Stupka and R. Ruppert are acknowledged for their participation in the early stages of the project.References
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 1–10. Search PubMed.
- (a) S. G. DiMagno, V. S.-Y. Lin and M. J. Therein, J. Am. Chem. Soc., 1993, 115, 2513 CrossRef CAS; (b) A. D. Hamilton, H.-D. Rubin and A. B. Bocarsly, J. Am. Chem. Soc., 1984, 106, 7255 CrossRef CAS; (c) J. A. Wytko, E. Graf and J. Weiss, J. Org. Chem., 1992, 57, 1016.
- L. D. Sarson, K. Ueda, M. Takeuchi and S. Shinkai, Chem. Commun., 1996, 619 RSC.
- C. Drexler, M. W. Hosseini, A. De Cian and J. Fischer, Tetrahedron Lett., 1997, 38, 2993 CrossRef CAS.
- (a) E. Graf, M. W. Hosseini and R. Ruppert, Tetrahedron Lett., 1994, 35, 7779 CrossRef CAS; (b) E. Graf, M. W. Hosseini, R. Ruppert, N. Kyritsakas, A. De Cian, J. Fischer, C. Estournès and F. Taulelle, Angew. Chem., Int. Ed. Engl., 1995, 34, 1115 CrossRef CAS; (c) F. Bockstahl, E. Graf, M. W. Hosseini, D. Suhr, A. De Cian and J. Fischer, Tetrahedron Lett., 1997, 38, 7539 CrossRef CAS.
- (a) F. L. Weitl and K. N. Raymond, J. Am. Chem. Soc., 1980, 102, 2289 CrossRef CAS; (b) K. N. Raymond, G. Müller and B. F. Matzanke, Top. Curr. Chem., 1984, 123, 49 Search PubMed.
- G. Mislin, E. Graf and M. W. Hosseini, Tetrahedron Lett., 1996, 37, 4503 CrossRef CAS.
- (a) L. K. Gottwald and E. F. Ullman, Tetrahedron Lett., 1969, 36, 3071 CrossRef; (b) F. A. Walker, Tetrahedron Lett., 1971, 52, 4949 CrossRef.
- J. P. Collman, R. R. Gagne, C. A. Reed, T. R. Halbert, G. Lang and W. T. Robinson, J. Am. Chem. Soc., 1975, 97, 1427 CrossRef CAS.
- C. Drexler, M. W. Hosseini, J.-M. Planeix, G. Stupka, A. De Cian and J. Fischer, Chem. Commun., 1998, 689 RSC.
- F. L. Weitl, K. N. Raymond, W. L. Smith and T. R. Howard, J. Am. Chem. Soc., 1978, 100, 1170 CrossRef CAS.
- J. S. Lindsey, J. Org. Chem., 1980, 45, 5215 CrossRef CAS.
- (a) E. Rose, M. Quelquejeu, C. Pochet, N. Julien, A. Kossanyi and L. Hamon, J. Org. Chem., 1993, 58, 5030 CrossRef CAS; (b) E. Rose, A. Lecas, M. Quelquejeu, A. Kossanyi and B. Boitrel, Coord. Chem. Rev., 1998, 178–180, 1407 CrossRef CAS.
- E. Rose, A. Cardon-Pilotaz, M. Quelquejeu, N. Bernard and A. Kossanyi, J. Org. Chem., 1995, 60, 3919 CrossRef CAS.
- (a) J. F. W. McOmie and M. L. Watts, Tetrahedron, 1968, 24, 2289 CrossRef CAS; (b) E. H. Vickery, L. F. Pahler and E. J. Eisenbraun, J. Org. Chem., 1979, 44, 4444 CrossRef CAS.
- R. A. Freitag and D. G. Whitten, J. Phys. Chem., 1983, 87, 3918 CrossRef CAS.
- (a) E. B. Fleischer and A. L. Stone, J. Am. Chem. Soc., 1968, 90, 2735 CrossRef CAS; (b) J. Wang Dirks, G. Underwood, J. Cox Matheson and D. Gust, J. Org. Chem., 1979, 44, 2551 CrossRef.
- R. A Freitag, J. A. Mercer-Smith and D. G. Whitten, J. Am. Chem. Soc., 1981, 103, 1226 CrossRef.
- M. J. Crossley, L. D. Field, A. J. Forster, M. M. Harding and S. Sternhell, J. Am. Chem. Soc., 1987, 109, 341 CrossRef CAS.
- T. Hayashi, T. Asai, F. M. Borgmeier, H. Hokazono and H. Ogoshi, Chem. Eur. J., 1998, 4, 1266 CrossRef CAS.
- J. Joffre and F. Joffre, Anacin, ver. 1.0, Ecole Nationale Supérieure de Chimie de Montpellier, Montpellier, France, 1996..
- K. Hatano, K. Anzai, T. Kubo and S. Tamai, Bull. Chem. Soc. Jpn., 1981, 54, 3518 CAS.
- OpenMolen, Interactive Structure Solution, Nonius B.V., Delft, The Netherlands, 1997..
Footnote |
| † Dedicated to H. B. Kagan on the occasion of his 71st birthday. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2002 |