A ceramic electrochemical microreactor for the methoxylation of methyl-2-furoate with direct mass spectrometry coupling
Virginie
Mengeaud
a,
Olivier
Bagel
a,
Rosaria
Ferrigno
a,
Hubert H.
Girault
*a and
Akhtar
Haider
b
aLaboratoire d’Electrochimie, Ecole Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland
bSochinaz, Rue principale, 1895 Vionnaz, Switzerland
First published on 22nd January 2002
Abstract
A ceramic electrochemical reactor (CEM) devoted to electrosyntheses was developed. The CEM was constituted by the assembly of five structured ceramic layers. On one layer, platinum interdigitated band electrodes with a submillimetre interelectrode gap were screen-printed. The microreactor chamber was constituted by seven channels and its volume was less than 100 µL. After a sintering step at 850 °C for 1 h, the CEM appeared as a solid and compact unit. The CEM was directly connected to the six port valve of a mass spectrometer allowing an on-line sampling and analysis of the reaction mixture. The methoxylation of the methyl-2-furoate was carried out and the effect of the residence time in the CEM was investigated thanks to mass spectrometry analyses.
I. Introduction
Miniaturised total analytical system (μTAS) is a particularly active and attractive field of research, mainly centred on the separation and characterisation of DNA, proteins and other biomolecules.1,2 The design of μTAS has required highly precise and sophisticated techniques,3 which were progressively and successfully transferred to the field of microreactors.4 Furthermore, the development of microreactors has logically taken benefits not only of the existing fabrication methods but also of the wide variety of materials already used, including silicon,5 glass4 or metals.6 The main interesting feature of microreactors comes from the decrease of one of the linear dimensions.4 This results in a high surface/volume ratio, which improves the mass and heat transfers. Both efficiency of separation and analysis time are improved3 and an increase in selectivity is generally observed. In the past decade, a broad variety of microreactors has been developed including micro heat exchangers,7 micromixers,5 integrated microreaction systems8 and electrochemical microreactors9,10 and, was successfully applied to different chemical reactions such as catalytic reactions,11 Ugi multicomponent reactions,12 Wittig reactions13 or peptide syntheses.14 Finally, recent works highlight the interest for the on-line coupling of microreactors with an analytical system, especially when microreactors are used for chemical production or mass screening.12 In this aim, mass spectrometry appears as a well-appropriate analytical technique, providing in the same time quantitative and qualitative data, notably structural information on both the products and intermediates.Among the various synthetic routes, the electrochemical approach offers two particular advantages. Firstly, electrochemical syntheses are generally performed at room temperature, which is crucial for an economic viewpoint. Secondly, no additional reactive intermediates are required in the medium and consequently, the reaction products are relatively pure. The formation of the active species takes place directly onto the electrode surface, which confers to the electroorganic reaction a great regio and stereo selectivity.15 However, the main drawback of the electrochemical approach arises from inhomogeneities of the electrical field and from energy losses due to the Joule heating effect between two distant electrodes. As shown in a previous study, the use of screen-printed interdigitated band electrodes with submillimetre interelectrode gap allowed a reduction of the ohmic loss up to 50%.16 Furthermore, this electrode configuration revealed to be well adapted to the electrosynthesis allowing successfully the methoxylation of furan,17 the epoxidation of propylene18 and the electrolysis of seawater.19 Owing to this particular geometry, the current efficiency was ranging from 68% to 87% and finally, the electrosyntheses were highly selective.
Taking into account the specific advantages of microreactors in terms of mass and heat transfers and the efficiency of the interdigitated electrodes configuration, a ceramic electrochemical microreactor (CEM) devoted to electrosyntheses was developed. In this paper, the CEM fabrication and its electrochemical behaviour are described. Five ceramic layers assembled under pressure and sintered at 850 °C for 1 h constituted the CEM. Interdigitated electrodes were screen-printed using a platinum ink and the microreactor chamber was composed of seven channels perpendicular to the electrodes. The CEM was directly coupled with the six port valve of a mass spectrometer allowing the on-line sampling and analysis of the reaction mixture. The electrochemical methoxylation of a furan derivative, namely the methyl-2-furoate, was carried out and the effect of the residence time in the CEM was optimised thanks to mass spectrometry analyses.
II. Experimental
II.1. Reagents and materials
Stock solutions of 10−5 M methyl-2-furoate (M2F, Fluka, Switzerland) and 10−5 M methyl-2,5-dihydro-2,5-dimethoxy-2-furancarboxylate (DMM2F, Across, Belgium) were prepared daily in 10−1 M sulfuric acid (Merck, Germany) in methanol (Fluka, Riedel-deHaën, Switzerland). In order to characterise the electrochemical behaviour of the screen-printed platinum electrodes in the CEM, a 10−4 M Fe(II) solution, with 10−1 M KCl (Fluka, Switzerland) as the supporting electrolyte, was prepared by dissolving the appropriate amount of potassium hexacyanoferrate(II) trihydrate (Fluka, Switzerland) in deionised water from a Milli-Q system (Millipore, Bedford, USA). The liquid sheath for mass spectrometry measurements was a mixture of methanol, water and acetic acid (Fluka) in the ratio 50∶49∶1 (v∶v∶v).Polydimethylsiloxane seals (PDMS, thickness: 2 mm) were prepared in plastic flasks by thoroughly mixing in the ratio 10∶1 (v∶v) the silicone elastomer base and the associated curing agent (Distrelec AG, Switzerland), followed by a thermal cure at 60 °C for 1 h. In order to prevent any chemical contamination, especially for mass spectrometry measurements, PTFE capillary tubing (external diameter: 1.9 mm, internal diameter: 1.0 mm) and nipples were used for all experiments (Metrohm, Switzerland).
II.2. The ceramic electrochemical microreactor (CEM) fabrication
In a single step, platinum interdigitated band electrodes (length = 29 mm, width = 1 mm) were screen-printed with a semi-automatic screen-printer (Dickfilm system AG, Switzerland) on a low-temperature co-fired ceramic soft tape, called Green Tape™ (80 mm × 50 mm × 0.254 mm). This Green Tape™ was mainly composed of aluminium borosilicate ceramic and polymer binders (Du Pont, Switzerland). The platinum ink (DC 430-79) was purchased from Heraeus (W.C. Heraeus, Hanau, Germany). The anode–cathode gap width was 500 μm. Anodes and cathodes were connected to two opposite lateral pads (pad length = 80 mm, pad width = 5 mm). After a 1 h drying period at 60 °C, a dielectric layer (GPA 96-115, W.C. Heraeus, Hanau, Germany) was spread manually over the two connecting pads, leaving a 15 cm2 working area composed of 40 platinum band electrodes (20 anodes + 20 cathodes). A second drying period was carried out in the same conditions (1 h, 60 °C). As shown in Fig. 1, five ceramic layers of the same external dimensions constituted the CEM. The microreactor chamber was composed of seven channels (50 mm × 1 mm × 0.254 mm) performed with a cutter in a ceramic tape. The latter was sandwiched between the screen-printed layer and a third layer with two small channels allowing the filling of the microreactor chamber. A top ceramic layer with two small apertures (inlet and outlet, diameter: 2 mm) was placed on the previous sheets. Finally, a fifth ceramic layer was deposited under the assembly to reinforce the CEM. The CEM was definitely assembled with a hydraulic press (250 kg cm−2) followed by a sintering step at 850 °C for 1 h.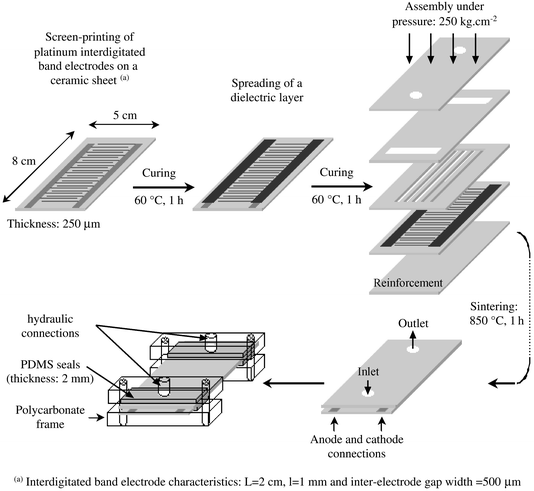
The last stage in the assembly procedure shown in Fig. 1 describes the connection between the capillary tubing and the CEM. Two PDMS seals with a 2 mm diameter hole in their centre were used directly on the inlet and outlet apertures of the CEM while two others were placed underneath the CEM. Both ends of the ensemble were clamped by polycarbonate blocks featuring a thread located directly above the inlet and outlet apertures. This arrangement ensures easy and leakage-free connections between the apertures of the CEM and the PTFE capillary tubing.
II.3. Instrumentations and electrochemical procedures
Mass spectrometry measurements were carried out on a Finnigan LCQ™DUO equipped with an electrospray ionisation source (ESI) and interfaced to a Gateway E-4200 PC system with Xcalibur™ software. The M2F stock solution was continuously delivered into the CEM by either a syringe pump (KD Scientific, Ismatec, Switzerland) or a peristaltic pump (Masterflex® Console drive, Cole-Parmer Instrument Company, USA).For the electrolysis of M2F, a Thurlby PL320QMD power supply (30V-2A, Thurlby Thandar Instruments, UK) was connected in a floating mode to the CEM. The electrochemical characterisation of the platinum band electrodes was carried out by cyclic voltammetry in a three-electrode configuration using a Portable Analytical Workstation (ECW2, Sycopel Scientific, UK). The connecting pads of the CEM were connected to the working electrode and the counter electrode plugs respectively. The pseudo-reference electrode (an Ag/AgCl wire) was located in one of the two apertures of the CEM.
II.4. ESI-MS and ESI-MS/MS
M2F and DMM2F were detected using electrospray ionisation in the positive mode. Data acquisition was performed in the full scan-mode from 50 to 190 m/z. The heated MS inlet capillary was held to 200 °C and the needle voltage was set to 4 kV. The solutions were nebulised by a coaxial flow of nitrogen through the spray needle at 20 arbitrary units (for a range from 20 to 100 arbitrary units), with a capillary voltage of 5.5 V. For all the experiments, the flow rate of the liquid sheath was 2.5 μL min−1.Two series of MS/MS experiments were performed. Solutions of M2F (10−5 M) and DMM2F (10−5 M) were analysed by infusion directly into the ESI interface at 2.5 μL min−1 in the liquid sheath. The mass range analysed varied between m/z 50 and 188 ([M + H]+ − 1). The ion energy of the first octapole was set to −4.2 V and −5.2 V, and the voltage of the second octapole was −7.5 V and −10.1 V, for M2F and DMM2F, respectively. The corresponding capillary voltages were 30 V and 5.5 V. In order to improve the signal-to-noise ratio of the mass spectral data, each scan was composed of 3 μscans (one mass analysis followed by ion detection). Ions were accumulated in the trap for up to 0.2 s. Helium was introduced as a collision gas into the ion trap at 1.6 mbar (1.22 × 10−5 Torr). The width of the mass window was set to m/z 1. The MS/MS spectra of both M2F and DMM2F were generated such that the amplitude of the rf voltage applied to the endcaps (normalised collision energy) was 30% and 32%, respectively.
III. Results and discussion
III.1. The ceramic electrochemical microreactor (CEM)
The fabrication of a microreactor usually requires sophisticated techniques,3 including wet chemical etching20 or laser ablation21 for the design of nano or micro volumes as well as for the introduction of electrodes in the reactor chambers. As shown in Fig. 1, a ceramic-based electrochemical reactor was fabricated involving three rapid and simple steps, namely screen-printing, sintering and curing steps. The based-ceramic sheets used were distinguished by both high flexibility and malleability. Consequently, the structuring of complex fluidic pattern can be easily envisaged. In addition, the softness of the ceramic sheets allowed a rapid formatting with a cutter of the different components of the CEM. The ceramic sheets also featured a smooth surface, which is well suited for the screen-printing of electrodes. Provided the matrix of the ink used resists to the high final temperature applied during the sintering step (850 °C), various kinds of electrodes such as platinum, gold or ruthenium oxide electrodes can be obtained in a reproducible manner. To that aim, an array of 40 platinum interdigitated band electrodes was screen-printed. In order to limit the ohmic loss, the inter-electrode gap width was 500 μm.16 The microchamber was constituted of seven channels of 254 μm depth, corresponding to an internal volume less than 100 μL. The sintering step ensured the definitive solidification of the CEM, leading to a solid and compact operational unit. As shown by the side view of the CEM (Fig. 2), an efficient sealing was obtained and no discrete ceramic layer was observable after the sintering step. This explains the efficient watertightness of the CEM. However, due to the assembly under-pressure, the depth of the channels decreased down to about 100 μm, reducing in the same time the internal volume. It is worth noting that the thickness could be increased using more than one spacing sheet. Finally, a slight retraction (10%) of the ceramic sheets occurred during the sintering step. Nevertheless, no structural deformation of the CEM was observable and no short circuit between anodes and cathodes were generated. After the sintering step, the smooth surface and the relative brittleness of the CEM avoided a direct connection to the CEM. In order to overcome this connecting difficulty, the CEM was maintained in a polycarbonate frame equipped with fluidic fittings. Two small PDMS seals ensured a watertight connection. The CEM appeared as a compact microreactor, which can be easily integrated in different places of a hydraulic system or connected to an analytical device such as a mass spectrometer. | ||
| Fig. 2 Side view of the ceramic electrochemical microreactor (CEM) composed of five sintered ceramic sheets. | ||
After the sintering step (850 °C for 1 h), the screen-printed electrodes were distinguished by an excellent adhesion and a high conductivity. The resistance value was closed to 10 Ω cm−1. The electrochemical behaviour of the platinum interdigitated band electrodes inside the CEM was verified by cyclic voltammetry with a 10−4 M Fe(II) test solution in 10−1 M KCl. The peak potential was independent of the scan rate value indicating a low ohmic loss in the CEM and the variation of the current density with the square root of the scan rate value was linear indicative of an electrochemical reaction controlled by a semi-infinite diffusion.22,23
The dimethoxylation of methyl-2-furoate (M2F),24 presented in Scheme 1, has been studied in the undivided CEM. Although the reaction mechanisms occurring at electrodes were not clearly identified, the anodic reaction could be preferentially assigned to the oxidation of methanol. Indeed, the product issued from the oxidation of methanol may activate M2F and consequently dimethoxylate the organic compound. The anodic methoxylation of M2F may correspond to a 2-electron process. The anodic reaction is accompanied by a hydrogen production at the cathode.
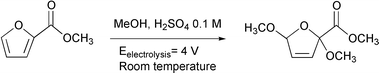 | ||
| Scheme 1 Electrochemical dimethoxylation of M2F. | ||
III.2. CEM-MS connecting set-up
The direct coupling of the CEM with a powerful analytical system, such as a mass spectrometer, represents an advantageous method to control both the reaction and the physical parameters of the CEM. Fig. 3 is a schematic representation of the connection of the CEM to the MS allowing the on-line analysis of the electrochemical synthesis. As represented in Fig. 3A, a syringe pump or a peristaltic pump, both ensuring a flow rate up to 250 μL min−1 for a single pass or a recirculation configuration respectively fed the CEM. The outlet of the CEM was directly connected to the injection valve of the mass spectrometer thanks to a tube ended by a needle (Unimetrics, Socochim/Alltech, Switzerland). The running of the six port valve allowing the on-line sampling of the reaction mixture is illustrated in Fig. 3B. In the load position, the reaction mixture coming from the CEM (port 1) was sent to either the waste or the peristaltic pump (port 2), passing continuously through the sample loop. In the inject position, the sample loop was connected to the liquid sheath flow (port 5) which pushed the sample into the ESI source of the mass spectrometer (port 4) while port 1 was directly connected to port 2 without passing through the sample loop. Consequently, a sampling of the reaction mixture can be easily achieved regardless of the hydrodynamic conditions and the flow configuration (single pass or recirculation). It is worth noting that feeding tubes and connectors were in PTFE, allowing a study in organic solvents without risk of contamination. In the same time, the CEM was connected to the power supply providing an electrolysis potential of 4 V. In order to prevent high voltage interferences between the ESI source and the power supply, the latter was operated in a floating potential configuration.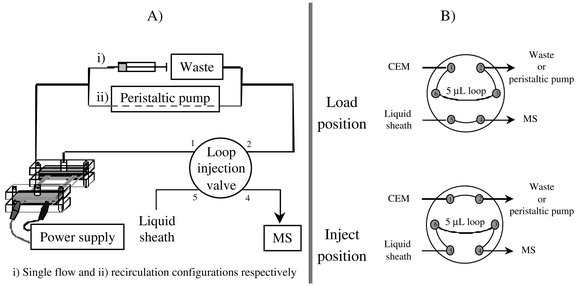 | ||
| Fig. 3 Schematic representation of the global set-up allowing the on-line mass spectrometry identification of the electro-generated product (A) and the two configurations of the six port valve (B). | ||
III.3. MS/MS characterisation of M2F and DMM2F
DMM2F was obtained by the electrochemical dimethoxylation of M2F in methanol with 10−1 M sulfuric acid,24 which served as the supporting electrolyte. Contrary to the methoxylation of furan,25 the methoxylation of M2F did not require any addition of salt in the medium. Consequently, this reaction can be followed by mass spectrometry without risks of perturbation of the MS signal due to blocking effects of ions, such as Na+ or K+, in the electrospray ionisation process.Fig. 4 shows the averaged daughter ion spectra (n = 130) of M2F and DMM2F, generated by collision-induced fragmentation in the positive ion mode. The ion abundance is represented in normalised scale where 1 equals 100% of the relative intensity. MS/MS fragmentation have been performed on the [M + H]+ parent ions at m/z 127 and 189 for M2F and DMM2F respectively. As portrayed in Fig. 4A, the fragmentation of M2F provided two main product ions. The most abundant corresponded to the [M − OCH3]+ ion at m /z 95 while the second fragment at m/z 109 resulted in the breaking of the carbonyl bond followed by a neutral fragment loss identified as a water molecule. Unfortunately, due to the instrumental range detection, the resulting methoxy fragment less than m/z 50 was not detected. In the same manner, Fig. 4B shows that the MS/MS fragmentation of DMM2F provided two main product ions. As for the M2F, the most abundant at m/z 157 was attributed to a methoxy loss. The second fragmentation of the DMM2F observed at m/z 129 came from the total cleavage of the ester function, namely [M − COOCH3]+. It appears that both M2F and DMM2F molecules cannot be further fragmented. The other product ions detected at m/z 55 in Fig. 4A and at m/z 147 and 165 in Fig. 4B may be attributed to recombination of different fragments.
![ESI-MS/MS averaged (n = 130) profile spectra with major fragments of methyl-2-furoate ([M + H]+ 127) and methyl-2,5-dihydro-2,5-dimethoxy-2-furancarboxylate ([M + H]+ 189). Samples were directly infused into the ESI interface at 2.5 μL min−1. Spectra were recorded in positive ion mode by scanning a mass range from m/z 50 to 188. Source conditions were needle voltage 4 kV, inlet capillary temperature 200 °C, capillary voltage: 30 V (A) and 5.5 V (B) and normalised collision energy: 30% (A) and 32% (B).](/image/article/2002/LC/b109587k/b109587k-f4.gif) | ||
| Fig. 4 ESI-MS/MS averaged (n = 130) profile spectra with major fragments of methyl-2-furoate ([M + H]+ 127) and methyl-2,5-dihydro-2,5-dimethoxy-2-furancarboxylate ([M + H]+ 189). Samples were directly infused into the ESI interface at 2.5 μL min−1. Spectra were recorded in positive ion mode by scanning a mass range from m/z 50 to 188. Source conditions were needle voltage 4 kV, inlet capillary temperature 200 °C, capillary voltage: 30 V (A) and 5.5 V (B) and normalised collision energy: 30% (A) and 32% (B). | ||
The ester function in both M2F and DMM2F gave rise to two different cleavages that can be explained considering the electronic distribution. The conjugation of three double bonds in M2F induced a homogeneous relocation of the π-electrons increasing the reactivity of the oxygen in the carbonyl group. After the methoxylation, this conjugation was not allowed any more and consequently the reactivity of this oxygen decreased. Clearly, M2F and DMM2F provided distinct MS signals allowing a rapid and easy identification of both. The progress of the electrochemical reaction can be envisaged following either the decrease of the most intense signal of M2F, namely the signal corresponding to the parent ion at m/z 127 or the increase of the product ion signal at m/z 157 which is the most abundant signal of DMM2F.
III.4. Direct coupling between the CEM and the mass spectrometer
In order to have a real-time mass identification of each compound present in the reaction mixture, the CEM was directly coupled to the mass spectrometer. Fig. 5 presents four averaged ESI mass spectra (n = 160) in normalised abundance scale obtained in the positive ion mode. The mass profile of the initial electrolytic solution containing 10−5 M M2F in 10−1 M H2SO4 in methanol and obtained for an applied potential of 0 V and for a flow rate of 250 μL min−1 is portrayed in Fig. 5A. Mass spectra of the electrolysed solution (the applied potential was 4 V) are presented in Fig. 5B, C and D for a flow rate of 250, 150 and 50 μL min−1 respectively.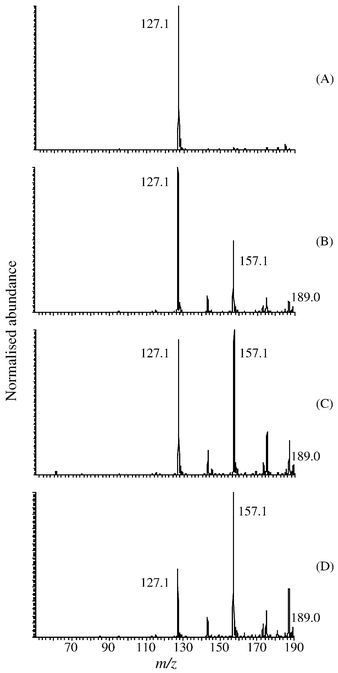 | ||
| Fig. 5 Electrospray ionisation averaged (n = 160) mass spectra in positive mode issued from a single-flow electrolysis of a mixture containing 10−5 M methyl-2-furoate and 0.1 M sulfuric acid in methanol at different flow rates: (B) 250, (C) 150 and (D) 50 μL min−1; (A) represented the initial solution. The liquid sheath was 50/49/1% methanol/water/acetic acid. | ||
As mentioned previously, the most intense signal of M2F was the signal corresponding to the [M + H]+ parent ion at m/z 127 (Fig. 5A). It is worth noting that the solution did not contain major impurities. The methoxylation of M2F occurred when the electrolysis potential (4 V) is applied to the CEM, resulting in the appearance of the product ion of DMM2F at m/z 157 as shown in Fig. 5B. The abundance of the [M + H]+ parent ion at m/z 189 distinguishing DMM2F was negligible. Fig. 5C and D shows the influence of the flow rate on the electrolysis efficiency. The decrease of the flow rate resulting in an increase of the residence time of the reaction mixture in the CEM allowed to inverse the ratio of the normalised abundance of M2F and DMM2F. Clearly, a low flow rate favoured the electrochemical methoxylation (Fig. 5D). The m/z 157 product ion of DMM2F represented the maximal abundance for a flow rate less than 150 μL min−1 while the [M + H]+ parent ion of the reactive M2F (at m/z 127) became minority.
At the same time, with the decrease of the flow rate, three other compounds appeared at m/z 145, 175 and 187. The m/z 187 ion was present at trace level in the mass spectrum of the commercial product DMM2F and could be a side-product of the electrosynthesis. The two other ions present in the mass spectra at m/z 175 and 145 were identified as the [M + H]+ parent ion and the [M − OCH3]+ product ion of the 2,5-dihydro-2,5-dimethoxy-2-furan carboxylic acid. Indeed, the acid derivative of DMM2F arises from the saponification equilibrium between the acid and its corresponding ester, which probably occurred at the same time as the electrosynthesis.
The electrosynthesis was accompanied by a cathodic hydrogen production in the CEM, which induces the formation of bubbles in the tubes. A quantitative sampling was delicate to achieve; the volume of the reaction mixture in the sample loop was not controlled. Consequently, without a downstream degassing device, a precise optimisation of either physical parameters of the CEM (length of the CEM, number of electrodes) or experimental electrolysis conditions (the applied potential) was difficult to achieve.
IV. Conclusion
A new electrochemical microreactor based on ceramic material has been designed and characterised. The particular property of the material provides a simple structuring method offering a strong sealing of the different layers of the cell. The design of the microsystem can be readily modified leading to complex fluidic structures corresponding to more specific applications. In addition, due to the inert property of the ceramic, electrochemical reactions can be carried out in a large class of organic solvents. Furthermore, such microdevices can be interfaced with a complete analytical technique such as GC-MS or HPLC-MS. Therefore, a mechanistic study of electrochemical reactions can be envisaged to identify intermediate species. Finally, future works may integrate the CEM in an operational single-flow system composed of the CEM feeding downstream chemical microreactors or other CEM ensuring a multi-step reaction.Acknowledgements
The authors acknowledge the Commission pour la Technologie et l’Innovation (C.T.I.) for financial support and are grateful to Du Pont de Nemours for their gift of Green Tape™.References
- J. Khandurina, T. E. Mcknight, S. C. Jacobson, L. C. Waters, R. S. Foote and J. M. Ramsey, Anal. Chem., 2000, 72, 2995 CrossRef CAS.
- G. H. W. Sanders and A. Manz, Trends Anal. Chem., 2000, 19, 364 CrossRef CAS.
-
W. Ehrfeld, V. Hessel and H. Löwe, Microreactors—New technology for modern chemistry,Wiley-VCH, Weinheim, 2000 Search PubMed.
![[*]](https://www.rsc.org/images/entities/char_e103.gif) Significant reference.
Significant reference. - S. J. Haswell, R. J. Middleton, B. O′sullivan, V. Skelton, P. Watts and P. Styring, Chem. Commun., 2001, 391 RSC.
- F. G. Bessoth, A. J. de Mello and A. Manz, Anal. Commun., 1999, 36, 213 RSC.
- W. Ehrfeld, K. Golbig, V. Hessel, H. Löwe and T. Richter, Ind. Eng. Chem. Res., 1999, 38, 1075 CrossRef CAS.
- J. Brandner, M. Fichtner and K. Schubert, Electrically heated microstructure heat exchangers and reactors, Microreaction Technology: 3rd International Conference on Microreaction Technology, Proceedings of IMRET 3,Frankfurt, 1999, p. 607 Search PubMed.
- H. Löwe and W. Ehrfeld, Electrochim. Acta, 1999, 44, 3679 CrossRef CAS.
- M. N. Mahmood and N. Bonanos, Solid State Ionics, 1992, 53–56, 142 CrossRef CAS.
- A. Ziogas, H. Löwe, M. Küpper and W. Ehrfeld, Elecrochemical microreactors: a new approach for microreaction technology, Microreaction Technology: 3rd International Conference on Microreaction Technology, Proceedings of IMRET 3,Frankfurt, 1999, p. 136 Search PubMed.
- N. G. Wilson and T. Mccreedy, Chem. Commun., 2000, 733 RSC.
-
M. C. Mitchell, V. Spikmans, A. Manz and A. J. de Mello, J. Chem. Soc., Perkin Trans. 1, 2001, 514 Search PubMed.
Significant reference.
- V. Skelton, G. M. Greenway, S. J. Haswell, P. Styring, D. O. Morgan, B. H. Warrington and S. Y. F. Wong, Analyst, 2000, 126, 7 Search PubMed.
- P. Watts, C. Wiles, S. J. Haswell, E. Pombo-Villar and P. Styring, Chem. Commun., 2001, 990 RSC.
- T. Shono, Electroorganic Synthesis, Academic Press, London, 1991 Search PubMed.
- C. Belmont and H. H. Girault, J. Appl. Electrochem., 1994, 24, 475 CrossRef CAS.
- C. Belmont and H. H. Girault, J. Appl. Electrochem., 1994, 24, 719 CrossRef CAS.
- C. Belmont and H. H. Girault, Electrochim. Acta, 1995, 40, 2505 CrossRef CAS.
- C. Belmont, R. Ferrigno, O. Leclerc and H. H. Girault, Electrochim. Acta, 1998, 44, 597 CrossRef CAS.
- T. R. Dietrich, W. Ehrfeld, M. Lacher, M. Krämer and B. Speit, Microelectron. Eng., 1996, 30, 497 Search PubMed.
- S. Zissi, A. Bertsch, J. Y. Jézéquel, S. Corbel and J. C. André, Microsystem Technol., 1996, 2, 97 Search PubMed.
- H. H. Girault, Electrochimie physique et analytique, Presses polytechniques et universitaire romandes,Lausanne, 2001 Search PubMed.
- J. S. Rossier, M. A. Roberts, R. Ferrigno and H. H. Girault, Anal. Chem., 1999, 71, 4294 CrossRef CAS.
- N. Clauson-Kaas, F. Limborg and K. Glens, Acta Chem. Scand., 1952, 6, 551 CAS.
- R. Ferrigno, V. Reid and H. H. Girault, Single flow electrochemical microreactor application to furan methoxylation, Microreaction Technology: 3rd International Conference on Microreaction Technology, Proceedings of IMRET 3, Frankfurt, 1999, p. 294 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |