Poly[bis(triphenylamine) ether]s with low glass transition temperatures as photoconductors in fast photorefractive systems
Jolita Ostrauskaite†a, Haridas R. Karickala, André Leopoldb, Dietrich Haarerb and Mukundan Thelakkat*a
aMakromolekulare Chemie I, University of Bayreuth, 95440, Bayreuth, Germany. E-mail: mukundan.thelakkat@uni-bayreuth.de
bExperimentalphysik IV, University of Bayreuth, 95440, Bayreuth, Germany
First published on 14th November 2001
Abstract
A series of photoconducting poly(tetraphenyldiaminobiphenylene alkyl ether)s in which tetraphenyldiaminobiphenyl (TPD) units are covalently linked through flexible oligomethylene glycol spacers in the main chain were synthesized and the thermal, optical and electrochemical properties were studied. Due to the introduction of flexible spacers, the polymers are highly soluble and could be obtained as film-forming materials with appreciably high molecular weights. The polymers exhibit glass transition temperatures between 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C and 128°C which is about 100°C less than those main chain polymeric bis(triphenylamine)s without such spacers. The HOMO value as determined from cyclic voltammetry is about −5.1 eV. The glass transition temperature of the photorefractive composites prepared by mixing the different polymers with an electro-optic chromophore, 1-(2-ethylhexyloxy)-2,5-dimethyl-4-(4-nitrophenylazo)benzene,
EHDNPB, could be tuned over a wide range about room temperature by just changing the photoconductor and without the need of any additional amount of plasticizer. Degenerate four-wave mixing and two-beam coupling in composites with the composition, photoconductor ∶ EHDNPB ∶ C60
(60 ∶ 39 ∶ 1 by wt/wt%) results in refractive index modulations of 10−3 with corresponding response time ∼10 ms and a photorefractive gain of Γ = 13 cm−1 for a writing beam intensity of 1 W cm−2
(645 nm) under an external electric field of 60 V µm−1.
°C and 128°C which is about 100°C less than those main chain polymeric bis(triphenylamine)s without such spacers. The HOMO value as determined from cyclic voltammetry is about −5.1 eV. The glass transition temperature of the photorefractive composites prepared by mixing the different polymers with an electro-optic chromophore, 1-(2-ethylhexyloxy)-2,5-dimethyl-4-(4-nitrophenylazo)benzene,
EHDNPB, could be tuned over a wide range about room temperature by just changing the photoconductor and without the need of any additional amount of plasticizer. Degenerate four-wave mixing and two-beam coupling in composites with the composition, photoconductor ∶ EHDNPB ∶ C60
(60 ∶ 39 ∶ 1 by wt/wt%) results in refractive index modulations of 10−3 with corresponding response time ∼10 ms and a photorefractive gain of Γ = 13 cm−1 for a writing beam intensity of 1 W cm−2
(645 nm) under an external electric field of 60 V µm−1.
Introduction
Since the first report of photorefractivity in polymeric materials in 19911 there has been an increasing amount of research activity in this field due to potential applications like holographic data storage,2 image processing,3 optical pattern recognition,2,4 medical imaging,5,6 novelty filtering7etc. The complex phenomenon of photorefractivity is defined as the local refractive index change in the bulk of the material due to an unsymmetrical charge distribution in the material caused by spatial variation of light intensity.8 This is realized by a combination of physical functions like light absorption followed by charge separation, trapping, charge distribution and build-up of internal space charge field under the influence of an external electric field. This requires properties like light sensitization, photoconduction, linear and non-linear electro-optical effects and charge trapping in the photorefractive system. The simple and most widely studied photorefractive systems consist of a polymeric photoconductor mixed with large amounts of chromophore, a very small amount of a sensitizer and varying amounts of a plasticizer to tune the glass transition temperature (Tg) of the composite to room temperature.9,10 However if the Tg of the photorefractive system is low, the chromophores reorient under the influence of the space charge field resulting in a large increase of refractive index change known as the orientational enhancement.11The most widely used photoconductor is polyvinylcarbazole (PVK) which has a Tg above 200°C. In sufficiently plasticized PVK-based photorefractive polymers, very large diffraction efficiencies, gain coefficients and fast response times have been observed.9,12,13 The main problem in these composites is the tendency of the electro-optic molecules to phase separate due to the non-compatibility of the photoconductor and chromophore.14 Another factor that has to be improved in such systems is the charge transport mobility, for PVK possesses relatively poor photoconductivity.15 It is also desirable to avoid the additional component of plasticizer by suitable structural variation of the photoconductor, to obtain low-Tg photorefractive systems.16,17 In order to improve the photoconductivity,
composites containing low molecular weight tetraphenyldiaminobiphenyl (TPD) derivatives,18,19 polymeric TPDs15,16 as well as copolymers carrying TPD units20 were examined, because TPD derivatives exhibit high hole transport mobility.
The problem of phase separation and the resulting instability of the system can be overcome by designing new photoconductors which are compatible in chemical structure with classical electro-optic chromophores. In this paper, the design, synthesis and characterization of a series of photoconducting polymers are described. In these polymers, highly photoconducting TPD units are covalently linked through flexible oligomethylene glycol spacers in the main chain. The synthetic strategy employed here allows the synthesis of low-Tg polymeric photoconductors with high content of the active TPD moiety. Photorefractive composites were prepared by blending the photoconductors with a highly soluble and compatible EO-chromophore carrying a branched alkoxy group, 1-(2-ethylhexyloxy)-2,5-dimethyl-4-(4-nitrophenylazo)benzene EHDNPB.21 The Tg of the photorefractive composites could be tuned over a wide range about room temperature by varying the photoconductor of the mixture and without using any additional amount of plasticizer. The advantage of this approach is that the amount of chromophore in the composite can be kept practically constant, during the variation of Tg. The photorefractive properties of some of these composites were studied using two-beam coupling and degenerate four-wave mixing experiments.
Results and discussion
Synthesis and characterization
The classical Ullmann reaction between a secondary diamine and a diiodide using copper catalysts at high temperatures of about 200°C is not suitable for the preparation of polymers due to the high amount of side reactions and the precipitation of oligomeric products due to insolubility. The modified reaction procedure using phase transfer catalysts reported by Frechet and Gauthier22 for low molecular weight compounds has been successfully utilized here for the preparation of soluble and high molecular weight triarylamine polymers. Three new bis(triphenylamine)-based polymers were synthesized as schematically shown in Fig. 1 from stoichiometric amounts of N,N′-diphenylbenzidine (1) and the corresponding bis(iodophenoxy)alkanes (2a–c) by Ullmann reaction using
Cu and K2CO3 in dry 1,2-dichlorobenzene in the presence of 18-crown-6 as phase transfer catalyst.23 Aryl diiodide monomers were designed to incorporate the oligomethylene glycol spacer groups. Bis(iodophenoxy)alkanes (2a–c) were prepared by the reaction between 4-iodophenol and the corresponding dibromoalkanes in the presence of potassium carbonate (see Experimental section). The poly[bis(triphenylamine) ether]s (3a–c) were subjected to Soxhlet extraction with ethanol and repeated reprecipitation from ethanol to remove the low molecular weight oligomeric fractions. In this way, high yields (above 80%) of high molecular weight polymers were obtained.![Scheme of synthesis of poly[bis(triphenylamine) ether]s 3a–c by Ullmann reaction.](/image/article/2002/JM/b105930k/b105930k-f1.gif) | ||
| Fig. 1 Scheme of synthesis of poly[bis(triphenylamine) ether]s 3a–c by Ullmann reaction. | ||
The incorporation of the oligomethylene glycol spacer into the main chain of the polymer, on the one hand, leads to increased solubility and, on the other hand, guarantees compatibility and therefore miscibility with chromophores carrying alkoxy spacers. On comparison, Ullmann polymerization with diiodides without spacers leads only to low molecular weight oligomeric mixture.24 The length of the oligomethylene glycol spacer group influences the thermal properties as well. The chromophore EHDNPB was prepared by a known procedure21 from 2,5-dimethyl-4-(4-nitrophenylazo)phenol by etherification with ethylhexyl bromide. The compound 2,5-dimethyl-4-(4-nitrophenylazo)phenol was prepared from 2,5-dimethylphenol and 4-nitrobenzenediazonium salt. All the materials synthesized were characterized by FTIR- and 1H-NMR spectroscopy and mass spectrometry measurements.
In the synthesis of polymers 3a–c, the strong IR absorption at 3380 cm−1 due to N–H stretching in compound 1 disappears completely and the characteristic C–N stretching at 1235–1256 cm−1 is observed in the products. Additionally, polymers 3a–c exhibit characteristic aromatic absorptions at 3035–3032 cm−1 (C–H stretching), at 1593–1591, 1509–1506, 1493–1488 cm−1 (C–C stretching), characteristic aliphatic absorption at 2937–2924, 2862–2853 cm−1 (C–H stretching) and C–O–C asymmetric stretching at 1250 ± 2 cm−1. The signals in the 1H NMR spectra of poly[bis(triphenylamine) ether]s 3a–c could be exactly assigned to the characteristic hydrogen atoms of these compounds. As expected, the signals of hydrogen atoms of the oligomethylene glycol chain are shifted gradually to the lower field and the methyleneoxy hydrogen (OCH2-) signals are observed at 3.95–3.90 ppm.
The average molecular weight and their distribution were detected by gel permeation chromatography (GPC) using polystyrene as standard. All the polymers exhibit appreciably high average molecular weights, Mn varying from about 8000 to 15000 and Mw values between 50000 and 60000 g mol−1
(see Table 1). In spite of the achieved high molecular weight, all the polymers are soluble in common solvents like THF, chloroform etc. It should be noted that these polymers form thin and stable amorphous films of high optical clarity from solution casting.
The thermal properties of polymers were examined by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) measurements. TGA measurement reveals that the polymers are highly thermally stable and the onset of decomposition occurs only above 410°C (Fig. 2). The polymers exhibit only a glass transition; 3a at 128°C, 3b at 102°C and 3c at 92°C and show no melting and no crystallization behavior on further cooling and heating cycles between −50°C and 250°C at 10 K min−1. The second heating curves of polymers are shown in Fig. 3. The polymeric bis(triphenylamine)s without any spacer show Tg above 200°C,23,25 and the introduction of a long oligomethylene glycol chain as spacer lowers the Tg value by more than 100°C (see Table 1). Thus, these photoconductors are suitable for the preparation of composites with Tg close to room temperature in order to exploit the orientation enhancement phenomenon.11
![Thermogravimetric analysis (TGA) curves of poly[bis(triphenylamine) ether]s 3a–c at heating rate of 10 K min−1 under N2 atmosphere.](/image/article/2002/JM/b105930k/b105930k-f2.gif) | ||
| Fig. 2 Thermogravimetric analysis (TGA) curves of poly[bis(triphenylamine) ether]s 3a–c at heating rate of 10 K min−1 under N2 atmosphere. | ||
![Differential scanning calorimetry (DSC) curves of poly[bis(triphenylamine) ether]s 3a–c
(2nd heating cycle at 10 K min−1).](/image/article/2002/JM/b105930k/b105930k-f3.gif) | ||
| Fig. 3 Differential scanning calorimetry (DSC) curves of poly[bis(triphenylamine) ether]s 3a–c (2nd heating cycle at 10 K min−1). | ||
Electrochemical and optical properties
The electrochemical stability and the reversibility of the redox process of the polymers 3a–c and the EO-chromophore were studied using cyclic voltammetry (CV). The measurements were carried out at a glassy carbon electrode in a solution of carefully dried acetonitrile containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) at room temperature. The potentials were measured against Ag/AgNO3 as reference electrode and each measurement was calibrated as usual with the standard ferrocene/ferrocenium (Fc) redox system.26 The HOMO and LUMO energy values of the compounds were determined from oxidation and reduction potentials respectively by taking the value of −4.8 eV as HOMO energy level for the Fc with respect to zero vacuum level as described by Daub et al. in the literature.27 The polymers are electrochemically stable, for the same redox potentials are observed for repeated cycles of redox processes. As expected for the bis(triphenylamine) unit, polymers 3a–c exhibit two oxidation steps corresponding to monocation and dication radicals. The first and second oxidation steps are completely reversible for the entire scan rate of 50 mV s−1 to 500 mV s−1.The cyclic voltammograms of the polymers measured at a scan rate of 50 mV s−1 are given in Fig. 4. Oxidation potentials and HOMO values determined from the first oxidation potentials with respect to ferrocene/ferrocenium as internal standard are presented in Table 2. These values (Eox1vs. Fc = 0.3 V and HOMO = −5.1 eV) for 3a–c are in the typical range of those for bis(triphenylamine) derivatives:23,24 the polymers do not show any reduction behaviour in the measurement range from +1.2 V to −2.3 V with respect to Fc. On the other hand, the EO-chromophore EHDNPB exhibits only a reversible reduction at −1.4 V vs. Fc corresponding to a LUMO value of −3.4 eV with respect to zero vacuum level. The measured LUMO-value is in agreement with the reported value in the literature28 and also with those for similar azo compounds.18
![Cyclic voltammetry curves of poly[bis(triphenylamine) ether]s 3a–c measured at 25 °C at a scan rate of 50 mV s−1vs.Ag/AgNO3 using ferrocene/ferrocenium as standard.](/image/article/2002/JM/b105930k/b105930k-f4.gif) | ||
| Fig. 4 Cyclic voltammetry curves of poly[bis(triphenylamine) ether]s 3a–c measured at 25°C at a scan rate of 50 mV s−1vs.Ag/AgNO3 using ferrocene/ferrocenium as standard. | ||
The optical properties of the polymers and photorefractive composites were investigated by measuring the UV–vis spectra of their solutions in CHCl3. It is very important to know the absorption spectra of polymers and photorefractive composites because it determines the wavelength region in which they can be used. It is important that the composite does not absorb in the photorefractive measurement region, for it can lead to competing non-photorefractive gratings. As expected, the polymers 3a–c exhibit very similar absorption spectra with two vibronic bands at 308 nm and 355 nm. The spectra are similar to the UV–vis absorption of the model compound, TPD. A comparison of the absorption spectra of the photoconductor 3a, composite (3a ∶ 3c ∶ EHDNPB ∶ C60 = 30 ∶ 30 ∶ 39 ∶ 1 wt/wt%) and the NLO-chromophore, EHDNPB, in CHCl3 is shown in Fig. 5. The composite shows a long-wavelength cut-off at 550 nm, making it possible to consider this material for photorefractive applications using laser light of 633 nm or 645 nm.
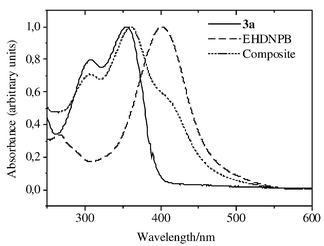 | ||
| Fig. 5 UV–vis spectra of 3a, EHDNPB and the composite containing 60% 3a ∶ 3c (1 ∶ 1)–39% EHDNPB–1% C60 in CHCl3 solution | ||
Photorefractive properties
Three photorefractive composites containing 60% of different photoconductors, 3a, 3a ∶ 3c (1 ∶ 1) and 3c respectively, 39% EHNDPB and 1% C60 as light sensitizer were prepared without adding any plasticizer. The thermal properties of these composites were characterized by DSC. All the composites show only a broad glass transition without any melting and recrystallization peaks, even though EHDNPB is a crystalline compound showing recrystallization and melting peaks in the second heating cycle at 27°C and 49°C respectively. The Tg values of the different composites with 60% of photoconductors, 3a, 3a
∶
3c
(1 ∶
1) and 3c are 42°C, 28°C and 12°C respectively. In this way we were able to tune the Tg of the composites over a range of temperatures about room temperature just by varying the composition of photoconductor and keeping the amount of EO-chromophore constant. Due to the difference in the length of the oligomethylene glycol spacers in the polymers, the effective wt% values of the TPD content in these three composites are different and are calculated to be 48.4%, 45.5% and 42.5% respectively. This fact should be taken into consideration along with the difference in Tg values in the discussion of photorefractive properties of the various composites. All these composites prepared with the poly[bis(triphenylamine) ether]s form optically clear films on filling cells of thickness between 25 and 80 µm and are stable for the last
one year. We reported earlier the applicability of low molecular weight triarylamine derivatives as photoconductors to obtain fast photorefractive systems having low Tg and containing no plasticizer.19 But the low molecular weight photoconductor system does not possess any such long-term stability due to phase separation of the components. The addition of plasticizer to lower the Tg and to stabilize the system has its own influence on the photorefractive effect.29 In order to understand the interplay of electrical and optical processes in photorefractive composites and the dynamics of the grating build-up, it is preferable to avoid the additional component of plasticizer and to have the minimum number of components in the composite. Thus the new polymers are very promising, for they can be blended with compatible NLO-chromophores to obtain low Tg composites which do
not show any phase separation.The photorefractive gain was measured using two-wave mixing experiments with p-polarized writing beams and calculated using the formula given in eqn. (1)
 | (1) |
 | (2) |
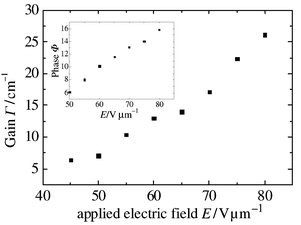 | ||
| Fig. 6 Photorefractive gain and phase dependence on the applied electric field for the composite, 3a ∶ 3c (1 ∶ 1)–39% EHDNPB–1% C60. | ||
To determine the response times of the material, we used a slight modification of the conventional biexponential fit, by replacing the fit to the slower component with a stretched exponential function, which provides more realistic data for chromophore orientation processes [eqn. (3)]:31
 | (3) |
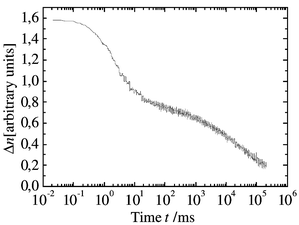 | ||
| Fig. 7 Example for an erasing process in degenerate four-wave mixing (DFWM) measurement with the composite, 60% 3a–39% EHDNPB–1% C60. | ||
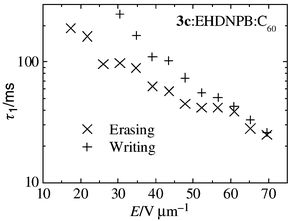 | ||
| Fig. 8 Dependence of photorefractive response times (fast component) on the applied electric field for the composite, 60% 3c–39% EHDNPB–1% C60. | ||
On comparing the different photorefractive composites, there is a clear difference in the response times as well as the achieved modulation of the refractive index. There is an order of magnitude difference between the fastest component of the two response times; the composite using polymer 3a being the fastest one (τ1 = 1 ms) and the composite using pure 3c being an order of magnitude slower (τ1 = 12 ms). The achieved refractive index modulations behave the opposite way: the material using 3c creates the highest Δn, which is explainable by the considerably lower Tg of the material, compared to the other composites29 (see Table 3).
| Photoconductor in composite/wt% | TPD content/wt% | Tg/°C | τ1/ms | Δn |
|---|---|---|---|---|
| 3a (60%) | 48.4 | 42 | 1 | 2 × 10−4 |
| 3a ∶ 3c (30% ∶ 30%) | 45.5 | 28 | 7 | 6 × 10−4 |
| 3c (60%) | 42.5 | 12 | 12 | 1 × 10−3 |
Ogino et al. have examined the photorefractive properties of composites prepared from copolymers carrying side chains of bis(triphenylamine) as well as butyl acrylate moieties and an EO-chromophore, DEANST, with C60 as sensitizer.15 Response times less than 10 ms were obtained at 50 V µm−1 for writing intensity of 250 mW cm−2. Therefore, on comparison the newly synthesized polymeric triarylamine ethers show very good photorefractive properties in composites with compatible NLO-dye.
Conclusion
We synthesized a series of polymeric bis(triphenylamine)s with ether linkages which are compatible with the NLO-dye, EHDNPB, so that different stable and low-Tg photorefractive composites could be prepared. The shelf-lives of these samples without any plasticizer are excellent. Two-beam coupling and four-wave mixing experiments were carried out to determine the photorefractive properties. The response time analysis indicates the two distinct processes due to the slow chromophore orientation and fast Pockels contribution. Fast response time of 1 ms was obtained for a composite with the photoconductor 3a. The low values of phase shift and gain exhibited by these composites are attributed to the presence of a cis–trans isomerization grating in this system.Experimental
Materials
All the chemicals were purchased from Aldrich as reagent grade and used as received without further purification, except N,N′-diphenylbenzidine (1), which was sublimed twice in vacuum before use. 1-(2-Ethylhexyloxy)-2,5-dimethyl-4-(p-nitrophenylazo)benzene (EHDNPB) was synthesized by a similar procedure to that described in literature.21 Solvents were distilled and dried as usual.Synthesis
°C) solution of 3.05 g (25 mmol) 2,5-dimethylphenol dissolved in 25 ml 10% NaOH solution, a cold solution of 5.92 g (25 mmol) of 4-nitrobenzenediazonium tetrafluoroborate dissolved in a mixture of 30 ml acetic acid and 30 ml water was added dropwise. The reaction mixture was stirred at 5°C for 1 h and it was kept alkaline throughout the reaction time by adding additional amounts of cold 10% NaOH solution if needed. The completion of reaction was tested by TLC (eluent: cyclohexane–ethyl acetate = 3 ∶ 1). After the reaction was completed, acetic acid was added to make the mixture acidic and the mixture was kept overnight in the fridge. The precipitated 2,5-dimethyl-4-(4-nitrophenylazo)phenol
was filtered, washed with water and dried. Yield = 6.4 g (94.4%) orange–red powder .Mp: 222–223°C (C14H13N3O3
= 271.15 g mol−1). IR (in KBr)
ν/cm−1: 3206 (O–H), 3069, 3013 (ar. C–H), 2923 (alk. C–H), 1591,1508 (ar. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C). MS (m/z): 271, 254, 241, 225, 195, 181, 149, 121, 92, 91, 77, 75. 1H-NMR (CDCl3), δ
(ppm): 2.31 (s, 3H, methyl), 2.72 (s, 3H, methyl), 5.12 (s, 1H, hydroxy), 6.78 (s, 1H, ar.), 7.65 (s, 1H, ar.), 7.98 (d, 2H, ar.), 8.38 (d, 2H, ar.).
C). MS (m/z): 271, 254, 241, 225, 195, 181, 149, 121, 92, 91, 77, 75. 1H-NMR (CDCl3), δ
(ppm): 2.31 (s, 3H, methyl), 2.72 (s, 3H, methyl), 5.12 (s, 1H, hydroxy), 6.78 (s, 1H, ar.), 7.65 (s, 1H, ar.), 7.98 (d, 2H, ar.), 8.38 (d, 2H, ar.).
4.07 g (15 mmol) of 2,5-dimethyl-4-(4-nitrophenylazo)phenol, 4.83 g (25 mmol) of ethylhexyl bromide, 3.45 g K2CO3 and a pinch of KI were added together in 100 ml of dry acetone and refluxed for 5 days. The mixture was filtered hot to remove the salts and the solvent was removed by rotovapor. The dark red oil obtained was dissolved in diethyl ether, washed with water to remove any inorganic impurities, dried over Na2SO4 and the ether was removed by rotovapor. The residue was recrystallized from methanol to obtain red needles of EHDNPB. Yield: 3.73 g (65%). Mp: 65°C (C22H29N3O3
= 383 g mol−1). IR (in KBr)
ν/cm−1: 2925, 1606, 1516, 1338, 1244, 1089, 859. 1H-NMR (CDCl3), δ
(ppm):
1.0 (m, 6H, CH3), 1.50 (m, 8H, CH2), 1.80 (m, 1H, CH), 2.30 (3H, CH3), 2.71 (3H, CH3), 3.92 (2H, OCH2), 6.75 (s, 1H, ar.), 7.62 (s, 1H, ar.), 7.94 (d, 2H, ar), 8.32 (d, 2H, ar). MS (m/z): 383 [M+], 271, 254, 149, 135, 121, 43.
Yield 9.6 g (80%) of 2a
(C18H20I2O2
= 522.12 g mol−1). Mp: 123–125°C. IR (in KBr)
ν/cm−1: 3086 (ar. C–H), 2937, 2862 (alk. C–H), 1584, 1490 (ar. CC), 1255 (C–O–C), 1063, 628 (C–I). 1H-NMR (CDCl3), δ
(ppm): 1.53 (m, 4H, alk.), 1.80 (t, 4H, alk.), 3.92 (t, 4H, alkoxy), 6.65 (m, 4H, ar.), 7.53 (m, 4H, ar.). MS (m/z): 522, 396, 303, 220, 203, 83.
°C. The mixture was allowed to warm to room temperature and was stirred until there was no more H2 evolution. Then 3.30 g (11.0 mmol) of 1,10-dibromodecane was added slowly. The mixture was heated to 120°C and stirred for 18 h, then cooled to room temperature. The solvent was removed by distillation. The crude product was dissolved in 100 ml CH2Cl2 and washed with 10% NaOH solution and then with water. The organic phase was dried over anhydrous Na2SO4, and the solvent was removed by rotary distillator. The product was purified by recrystallization from isopropyl alcohol and then from n-hexane.Yield: 3.25 g (51.1 %) of 2b
(C22H28I2O2
= 578.23 g mol−1). Mp: 109–110°C. IR (in KBr)
ν/cm−1: 3063 (ar. C–H), 2928, 2851 (alk. C–H), 1570, 1488 (ar. CC), 1248 (C–O–C). MS (m/z): 578, 452, 220. 1H-NMR (CDCl3), δ
(ppm): 1.39–1.45 (m, 12H, alk.), 1.77 (q, 4H, alk.), 3.89 (t, 4H, alkoxy), 6.66 (d, 4H, ar.), 7.53 (d, 4H, ar.).
°C IR (in KBr)
ν/cm−1: 3081 (ar. C–H), 2938, 2850 (alk. C–H), 1586, 1487 (ar. CC), 1253 (C–O–C), 1056, 624 (C–I). MS (m/z): 606, 480, 386, 220, 186, 69, 55. 1H-NMR (CDCl3), δ
(ppm): 1.29 (s, 16H, alk.), 1.75 (s, 4H, alk.), 3.90 (s, 4H, alkoxy), 6.65 (s, 4H, ar.), 7.51 (s,
4H, ar.).Yield: 6.6 g (90%). IR (in KBr)
ν/cm−1: 3035 (ar. C–H), 2937, 2862 (alk. C–H), 1593, 1507, 1494 (ar. CC), 1272 (C–N), 1256 (C–O–C). UV (in CHCl3)
λ/nm: 308, 355. 1H-NMR (CDCl3), δ
(ppm): 1.55 (s, 4H, alk.), 1.81 (s, 4H, alk.), 3.95 (s, 4H, alkoxy), 6.6–7.6 (m, 26H, ar.).
C), 1272 (C–N), 1235 (C–O–C). UV (in CHCl3)
λ/nm : 308, 355. 1H-NMR (CDCl3), δ
(ppm): 1.3–1.67 (m, 12H, alk.), 1.78 (s, 4H, alk.), 3.90 (s, 4H, alkoxy), 6.7–7.6 (m, 26H, ar.).C), 1276 (C–N), 1238 (C–O–C). UV (in CHCl3)
λ/nm: 308, 355. 1H-NMR (CDCl3), δ
(ppm): 1.40 (d, 16H, alk.), 1.77 (s, 4H, alk.), 3.92 (t, 4H, alkoxy), 6.6–7.6 (m, 26H, ar.).Measurement
Nuclear magnetic resonance (1H-NMR) spectra were measured using a Bruker AC 250(250 Hz), IR absorption spectra using a Bio-Rad Digilab FTS-40 and UV–vis absorption spectra using a Hitachi U-3000 spectrophotometer. Mass spectra were obtained on a Finnigan MAT 8500 (70 eV) with a MAT 112 S Varian. Differential scanning calorimetry (DSC) measurements were carried out with a Perkin-Elmer DSC-7 at a heating rate of 10 K min−1 under N2 atmosphere. Thermogravimetric analysis was performed on a Netzch STA 409 with a data acquisition system 414/1 at 10 K min−1 heating rate under N2 atmosphere. Cyclic voltammetry (CV) measurements were done at a glassy carbon working electrode in a three-electrode and potentiostat configuration from EG&G Princeton Applied Research. Molecular weight of polymers was determined by gel permeation chromatography (GPC) using Waters HPLC (PS standard).Sample preparation
The photorefractive measurements were performed on films with thicknesses between 20 and 80 µm. The composites were prepared by mixing solutions containing 60 wt% of the photoconductor, 39 wt% of the chromophore EHDNPB and 1 wt% of C60 in benzene followed by filtration through a 0.2 µm filter and finally freeze-drying overnight under vacuum. For preparation of the photorefractive samples, the composite material was put onto a glass plate partially covered with indium tin oxide (ITO) and subsequently heated to 160°C. At this temperature, a second ITO covered glass plate was put on top of the first one. Spacers with a definite thickness were put in between the two glass plates to maintain the desired thickness of the photorefractive samples.Photorefractive measurements
The photorefractive measurements were performed using four-wave mixing or two-beam coupling experiments. A coherent 899 dye laser, operating at a wavelength of 645 nm was used for the writing beams, meeting the sample with an external angle of 20° in between them. The sample itself was tilted by 60°. For the four-wave mixing (FWM) experiments, a second, p-polarized laser beam at 670 nm, generated by a laser diode, counterpropagated one of the s-polarized writing beams. Photodiodes (Thor Labs PDA55EC) attached to lock-in amplifiers (Stanford SR830) were used to detect the diffracted and transmitted reading laser. FWM writing processes were performed switching on the second writing beam using a fast (400 µs) mechanical shutter, while the first one illuminated the sample previously. The erasure of the photorefractive grating was done by blocking the writing beam again, leaving the second one illuminating the sample.Acknowledgements
We thank Professor J. V. Grazulevicius (Kaunas University of Technology, Lithuania) and Professor H.-W. Schmidt (University of Bayreuth, Germany) for providing JO with a stipend for the period of stay at Bayreuth, Mrs A. Lang and Mrs H. Wietasch for their help in sample preparation and synthesis. Financial support from Bayerischer Forschungsstiftung, Projekt: “Organische optische Funktionsmaterialien” is kindly acknowledged.References
- S. Ducharme, J. C. Scott, R. J. Twieg and W. E. Moerner, Phys. Rev. Lett., 1991, 66, 1846 CrossRef CAS.
- I. McMichael, W. Christian, D. Pletcher, T. Y. Chang and J. H. Hong, Appl. Opt., 1996, 35, 2375 Search PubMed.
- C. Denz, T. Dellwig, J. Lembcke and T. Tschudi, Opt. Lett., 1996, 21, 278 Search PubMed.
- B. L. Volodin, B. Kippelen, K. Meerholz, B. Javidi and N. Peyghambarian, Nature, 1996, 383, 58 CrossRef CAS.
- S. C. W. Hyde, N. P. Barry, R. Jones, J. C. Dainty, P. M. W. French, M. B. Klein and B. A. Wechsler, Opt. Lett., 1995, 20, 1331 Search PubMed.
- D. D. Steele, B. L. Volodin, O. Savina, B. Kippelen, N. Peyghambarian, H. Röckel and S. R. Marder, Opt. Lett., 1998, 23, 153 Search PubMed.
- A. Goonesekera, D. Wright and W. E. Moerner, Appl. Phys. Lett., 2000, 76, 3358 CrossRef CAS.
- D. D. Nolte, Photorefractive effects and materials, Kluwer Academic PublishersLondon, 1995 Search PubMed.
- W. E. Moerner and S. M. Silence, Chem. Rev., 1994, 94, 127 CrossRef CAS.
- S. J. Zilker, ChemPhysChem, 2000, 1, 72 CrossRef CAS.
- W. E. Moerner, S. M. Silence, F. Hache and G. C. Bjorklund, J. Opt. Soc. Am. B., 1994, 11, 320 Search PubMed.
- M. A. Diaz-Garcia, D. Wright, J. D. Casperson, B. Smith, E. Glazer and W. E. Moerner, Chem. Mater., 1999, 11, 1784 CrossRef CAS.
- K. Meerholz, B. L. Volodin, Sandalphon, B. Kippelen and N. Peyghambarian, Nature, 1994, 371, 497 CrossRef CAS.
- F. Würthner, R. Wortmann, R. Matschiner, K. Lukaszuk, K. Meerholz, Y. De Nardin, R. Bittner, C. Bräuchle and R. Sens, Angew. Chem., Int. Ed. Engl., 1997, 36, 2765 CrossRef CAS.
- K. Ogino, T. Nomura, T. Shichi, S.-H. Park and H. Sato, Chem. Mater., 1997, 9, 2768 CrossRef CAS.
- H. J. Bolink, V. V. Kasnikov, P. H. J. Kouwer and G. Hadziioannou, Chem. Mater., 1998, 10, 3951 CrossRef CAS.
- S. Schloter, U. Hofmann, P. Strohriegl, H.-W. Schmidt and D. Haarer, J. Opt. Soc. Am. B, 1998, 15(9), 2473 Search PubMed.
- M. Thelakkat, C. Schmitz, C. Hohle, P. Strohriegl, H.-W. Schmidt, U. Hofmann, S. Schloter and D. Haarer, Phys. Chem. Chem. Phys., 1999, 1(8), 1693 RSC.
- U. Hofmann, S. Schloter, A. Schreiber, K. Hoechstetter, G. Bauml, S. J. Zilker, D. Haarer, M. Thelakkat, H.-W. Schmidt, K. Ewert and C.-D. Eisenbach, Proc. SPIE – Int. Soc. Opt. Eng., 1998, 3471, 124 Search PubMed.
- U. Hofmann, A. Schreiber, D. Haarer, S. J. Zilker, A. Bacher, D. D. C. Bradley, M. Redecker, M. Inbasekaran, W. W. Wu and E. P. Woo, Chem. Phys. Lett., 1999, 311, 41 CrossRef CAS.
- A. M. Cox, R. D. Blackburn, D. P. West, T. A. King, F. A. wade and D. A. Leigh, Appl. Phys. Lett., 1996, 68, 2801 CrossRef CAS.
- S. Gauthier and J. M. J. Frechet, Synthesis, 1987, 383 CrossRef CAS.
- M. Thelakkat, C. Schmitz and H.-W. Schmidt, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1999, 40(2), 1230 Search PubMed.
- M. Thelakkat, R. Fink, F. Haubner and H.-W. Schmidt, Macromol. Symp., 1997, 125, 157 Search PubMed.
- M. Thelakkat, J. Hagen, D. Haarer and H.-W. Schmidt, Synth. Met., 1999, 102, 125 CrossRef.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 462.
- J. Pommerehne, H. Vestweber, W. Gusws, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 55 CrossRef CAS.
- D. P. West, M. D. Rahn, C. Im and H. Bässler, Chem. Phys. Lett., 2000, 326, 407 CrossRef CAS.
- A. Leopold, U. Hofmann, M. Grasruck, S. J. Zilker, D. Haarer, J. Ostrauskaite, J. V. Grazulevicius, M. Thelakkat, C. Hohle, P. Strohriegl, H.-W. Schmidt, A. Bacher, D. D. C. Bradley, M. Redecker, K. Inbasekharan, W. W. Wu and E. P. Woo, Proc. SPIE – Int. Soc. Opt. Eng., 2000, 4104, 95 Search PubMed.
- C. A. Walsh and W. E. Moerner, J. Opt. Soc. Am. B, 1992, 9, 1642 Search PubMed.
- G. Williams and D. C. Watts, Trans. Faraday Soc., 1970, 66(1), 80 RSC.
Footnote |
| † Permanent address: Department of Organic Technology, Kaunas University of Technology, 3028 Kaunas, Lithuania. |
| This journal is © The Royal Society of Chemistry 2002 |