Synthesis and properties of weakly coupled dendrimeric multiporphyrin light-harvesting arrays and hole-storage reservoirs†
María
del Rosario Benites
a,
Thomas E.
Johnson‡
a,
Steven
Weghorn
a,
Lianhe
Yu
a,
Polisetti Dharma
Rao
a,
James R.
Diers
b,
Sung Ik
Yang
c,
Christine
Kirmaier
c,
David F.
Bocian
*b,
Dewey
Holten
*c and
Jonathan S.
Lindsey
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina 27695-8204, USA
bDepartment of Chemistry, University of California, Riverside, California 92521-0403, USA
cDepartment of Chemistry, Washington University, St. Louis, Missouri 63130-4889, USA
First published on 15th November 2001
Abstract
A convergent synthesis employing porphyrin building blocks has afforded dendrimeric multiporphyrin arrays containing n Zn-porphyrins (n = 4, 8, or 20) and one free base- (Fb-) porphyrin joined via diarylethyne linkers. Size exclusion chromatography was used extensively for purification. The arrays have sufficient solubility in toluene or other solvents for routine handling. With increasing size, the intense near-UV Soret (S0 → S2) absorption band broadens, splits, and red shifts due to inter-porphyrin exciton coupling. In contrast, the weaker visible bands (S0 → S1) remain essentially unchanged in position or width in proceeding from the monomer all the way to the 21-mer; however, the molecular extinction coefficients of the visible bands scale with the number of porphyrins. Similarly, the one-electron oxidation potentials of the porphyrins are virtually unchanged as the arrays get larger. These results are indicative of relatively weak (but significant) electronic coupling between ground states and between the photophysically relevant lowest-excited-singlet states of the diarylethyne-linked porphyrins; thus, the characteristic properties of the individual units are retained as the architectures increase in complexity. Efficient excited-singlet-state energy transfer occurs among the Zn-porphyrins and ultimately to the sole Fb-porphyrin in each of the arrays, with the overall arrival time of energy at the trapping site increasing modestly with the number of Zn-porphyrins = 1 (45 ps), 2 (90 ps), 8 (105 ps), and 20 (220 ps). The overall energy-transfer efficiencies are 98%, 96%, 96%, and 92% in the same series. The ground-state hole-storage properties of the 21-mer (Zn20Fb) were examined. Bulk electrolysis indicates that 21 (or more) electrons can be removed from this array (e.g., one hole resides on each porphyrin) to yield a stable “super-charged” π-cation radical. Taken together, these results indicate that the convergent building-block synthesis approach affords dendrimeric multiporphyrin arrays with favorable properties for light-harvesting and hole storage.
Introduction
As part of a program in artificial photosynthesis, we have been working on the design, synthesis, and characterization of multiporphyrin light-harvesting arrays.1 Synthetic multiporphyrin arrays are useful models of natural photosynthetic antennas and also provide the basis for a variety of molecular photonic devices, including sensors, nanoscale optical sources, and solar collectors. An effective light-harvesting array absorbs intensely and transfers the resulting electronic excited-state energy to one site with high efficiency. Accordingly, the strong light-absorbing properties of porphyrins make these macrocycles ideal for light-harvesting applications.2 Multiporphyrin arrays can also function as nanoscale hole/electron storage reservoirs because porphyrins form relatively stable π-cation radicals. Thus, a multiporphyrin construct affords the possibility of storing large numbers of electron equivalents.3,4Two basic types of approaches have been taken in the design of multi-chromophoric arrays for light-harvesting applications. Both approaches have favorable characteristics depending on the desired properties, design constraints, and assembly strategies. One approach employs linkers that give very strong electronic interactions between the units.5–11 In the strong coupling limit, such constructs typically exhibit supermolecular properties in that energy/electron delocalization involves wavefunctions that extend over a large number of chromophores. Consequently, such arrays have many characteristics that are much different from those of the isolated pigments, and that can change substantially with the size and architecture of the assembly. The second approach employs linkers that give rather modest couplings between pigments: such couplings are sufficiently weak so that key properties (e.g., ground-state redox potentials, photophysical characteristics of the lowest excited singlet state) designed into the individual pigments are essentially retained in the arrays, yet are of sufficient magnitude to support ultrafast excited-state energy transfer and facile ground-state hole/electron hopping. We have chosen this second strategy and have found that the diarylethyne linker affords the desired inter-pigment couplings, molecular properties, and wide versatility in a molecular building-block approach.2–4,12–23
Several distinct routes have been reported for synthesizing multiporphyrin arrays with potential light-harvesting properties. Self-assembly routes have afforded windowpane arrays of Pd-coordinated pyridyl-substituted porphyrins,24 indefinite aggregates of Zn-chlorins bearing hydroxy and keto groups,25 discrete aggregates of pyridyl-coordinated Zn-porphyrins,26 and other diverse architectures.27Polymerization approaches have yielded backbone polymeric arrays of up to 128 porphyrins with direct meso–meso linkages,9 or shorter polymers with oligo(phenylenevinylene),28 phenylethyne,29 or butadiyne linkers.30Stepwise synthesis procedures have afforded the majority of synthetic light-harvesting arrays. The chemistries employed have included (1) Wittig reactions affording stilbene linkers;31 (2) Sonogashira reactions affording diphenylethyne,2,12–23 oligophenylethyne,32 or diethynylethene linkers;33 (3) Glaser reactions affording diphenylbutadiyne linkers;23,34 (4) Heck reactions yielding ethene linkers;35 (5) condensation reactions affording phenylene linkers;36,37 (6) alkylation reactions affording benzoxyphenyl linkers,38 polyalkoxy linkers39 or 1,3,5-triazine units joining aniline groups;40 (7) amide forming reactions,41,42 and (8) a sequence of complementary chemistries including the Sonogashira reaction affording diphenylethyne and other linkers.17,43,44 It is noteworthy that some routes have led to porphyrin dendrimers. In particular, dendrimers have been prepared that are composed of one porphyrin at the core;45 16,41 32,42 or 64,41 porphyrins at the periphery; or up to 21 porphyrins in the framework of the dendrimer.37
We have employed the Sonogashira reaction46 in a building block approach for the modular construction of covalently linked multiporphyrin arrays.2,23 This modular route takes advantage of the availability of porphyrin building blocks with control over the pattern of the substituents at the four meso-positions. The typical substituents are aryl rings that bear solubilizing groups (e.g., mesityl) and/or synthetic handles (e.g., iodo and ethynyl groups) for further elaboration. The porphyrins are used as the free base (Fb) or metal chelate. The Pd-coupling reactions of iodophenyl and ethynylphenyl porphyrin building blocks yield diarylethyne linkages and are performed under mild, non-acidic, non-metalating conditions, which preserve the metalation state of the porphyrin building blocks.47,48 The diarylethyne linker undergoes a modest degree of bending49 and enables free rotation (about the cylindrically symmetric ethyne unit), thereby affording a relatively fixed distance of separation and access to all dihedral angles of adjacent linked porphyrins.50 We have employed this synthetic approach to construct arrays consisting of porphyrins or porphyrins plus accessory pigments in linear,2,13,21 T-shaped,51 star-shaped,12,19 cyclic,16 or square15 architectures. The constructs include light-harvesting arrays, optoelectronic gates, and a variety of dyads and triads to probe the mechanisms of excited-state energy transfer and ground-state hole/electron hopping between porphyrins and between porphyrins and accessory chromophores. Each of our diarylethyne-linked arrays prepared and studied to date has been comprised of six or fewer porphyrins. A selection of such arrays includes dimers (ZnFbU,23 ZnFbP,2 Zn2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23), molecular squares (cyclo-Zn2Fb2U, cyclo-Zn4U),15 trimers (Zn3, ZnZnFb),2 and star-shaped pentamers (Zn4FbU, Zn5U)12,19 as shown in Charts 1 and 2;52 the properties of these arrays provide the benchmarks for understanding the properties of the new arrays described herein.
23), molecular squares (cyclo-Zn2Fb2U, cyclo-Zn4U),15 trimers (Zn3, ZnZnFb),2 and star-shaped pentamers (Zn4FbU, Zn5U)12,19 as shown in Charts 1 and 2;52 the properties of these arrays provide the benchmarks for understanding the properties of the new arrays described herein.
 | ||
| Chart 1 Representative arrays prepared previously.50,52 | ||
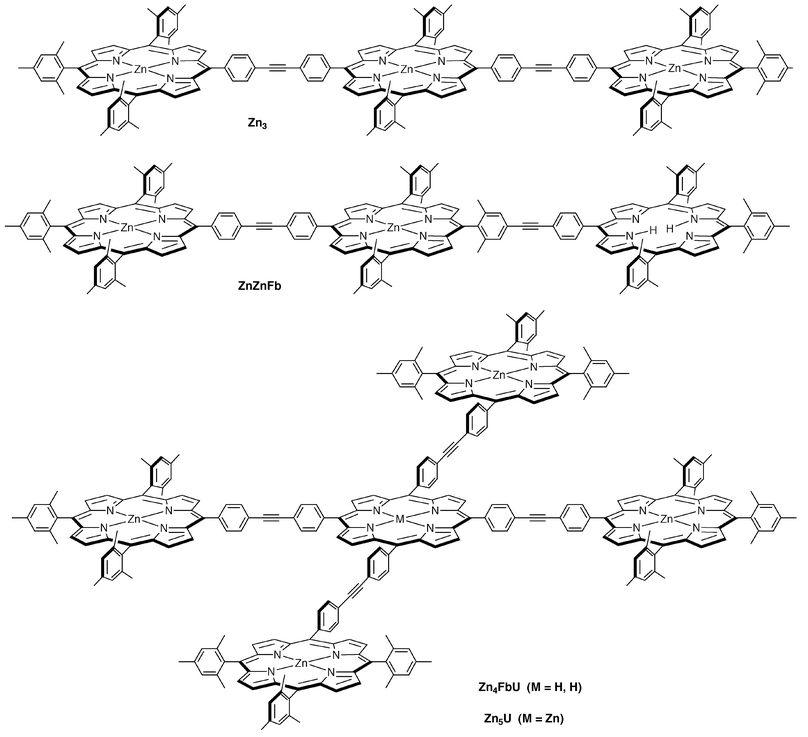 | ||
| Chart 2 Representative arrays prepared previously.50,52 | ||
In this paper, we have extended the building block approach to the synthesis of dendrimeric multiporphyrin arrays containing up to five, eight, nine, or 21 porphyrins. The arrays contain Zn-porphyrins and one (or no) Fb-porphyrin and are joined via diarylethyne linkers. Issues related to synthesis include the size of the array that can be constructed, the solubility of the larger arrays, and the use of size exclusion chromatography (SEC) as a means of purification. The arrays have been characterized by static and time-resolved optical spectroscopy to investigate their light-harvesting and energy-funneling characteristics, and electrochemistry to probe their hole-storage capabilities.
Results and discussion
1. Molecular design
Our objective was to prepare a family of dendrimeric arrays in which a single Fb-porphyrin resides at the core of the assembly and a variable number of Zn-porphyrins project outward from the core. The overall architecture depends on (1) the design of the Fb-porphyrin building block (which influences the shape of the array via the directions in which the Zn-porphyrin-containing antenna unit is attached) and (2) the number of generations of Zn-porphyrins (which influences the size of the array). The diarylethyne linker that joins the porphyrins is semi-rigid49 and allows significant electronic communication between the porphyrins.3,4,20,53 The non-linking meso-positions are substituted with mesityl groups to engender solubility in organic solvents.2. Synthesis
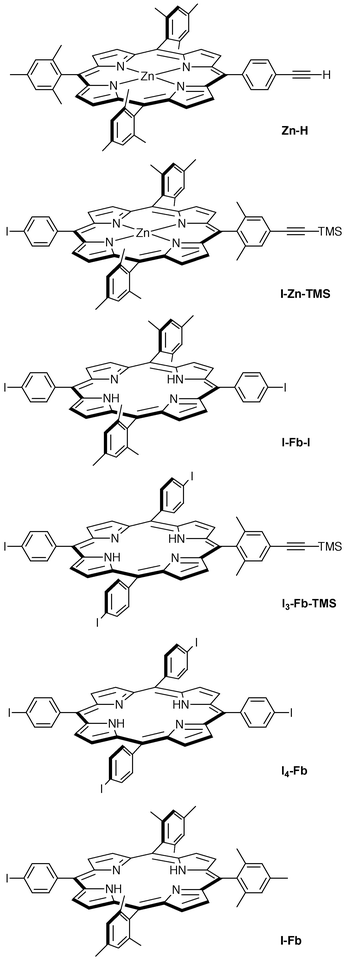 | ||
| Chart 3 Monomeric porphyrin building blocks. | ||
(a) Tetramers. The reaction of I3-Fb-TMS and Zn-H was performed using standard Pd-mediated coupling conditions47 (Scheme 1). These conditions include ∼2.5 mM concentrations of reactants with Pd2(dba)3 and AsPh3 in toluene/triethylamine at 35
°C under Ar in the dark. These conditions avoid the use of copper reagents, afford coupling in dilute solution, and do not alter the metalation states of the porphyrin building blocks. The reaction was monitored by analytical SEC.22,47 After 20 h, the analytical SEC showed four peaks consisting of the following materials (absorption data uncorrected for extinction coefficients): higher molecular weight material (HMWM, 5%), desired tetramer and other unidentified products (75%), dimeric product (3%), and unidentified monomeric porphyrins (17%). Purification by chromatography (silica and preparative SEC) afforded the Zn3Fb-TMS tetramer in 61% isolated yield. Metalation of Zn3Fb-TMS with Zn(OAc)2·2H2O afforded the all-Zn-tetramer Zn4-TMS in 92% yield. Treatment of Zn4-TMS with Bu4NF in THF for 1 h at room temperature gave the monoethyne-substituted tetramer Zn4-H (94%). The arrays Zn3Fb-TMS, Zn4-TMS and Zn4-H provided satisfactory absorption, 1H NMR, and laser-desorption mass spectrometry (LD-MS) data. The constitution of Zn4-H was further confirmed by high-resolution mass spectrometry (HRMS) analysis. The tetramer Zn4-H was employed in a molecular dynamics study using 1H NMR spectroscopy to assess the motional freedom of the linker (rotation and bending),49 and as the antenna component of integrated antenna-reaction center complexes.18 The tetramer Zn4-H also was converted to a diphenylbutadiyne-linked Zn8 octamer via Glaser oxidative homocoupling (Electronic Supplementary Information†). The Zn8 octamer lacks a Fb-porphyrin to serve as a low-energy trapping site for light-harvesting applications.
 | ||
| Scheme 1 Synthesis of tetrameric arrays.50 | ||
(b) Asymmetric pentamer Zn4Fb. A star-shaped pentamer was prepared which differs from the star-shaped pentamers that we have prepared previously. The latter have 4-fold symmetry with a core Fb-porphyrin and four identical metalloporphyrins at the periphery of the array. In contrast, the asymmetric Zn4Fb pentamer incorporates the Fb-porphyrin at the periphery of the array (Scheme 2). This light-harvesting array was prepared in 81% yield by the Pd-mediated coupling of the mono-ethynyl tetramer Zn4-H with a mono-iodo Fb-porphyrin (I-Fb). Analysis by absorption spectroscopy, 1H NMR spectroscopy, and LD-MS afforded satisfactory data.
 | ||
| Scheme 2 Synthesis of the asymmetric Zn4Fb pentamer.50 | ||
(c) Nonamer Zn8Fb. Tetramer Zn4-H also served as a monoethynyl building block in the convergent synthesis of a nonameric array. Thus, coupling of the diiodo Fb-porphyrin I-Fb-I and Zn4-H for 19 h afforded nonamer Zn8Fb (Scheme 3). In this array, the core porphyrin is surrounded by two tetrameric porphyrin units trans to each other. Analytical SEC of the reaction mixture showed 90% conversion of the starting porphyrins to the nonamer, as well as some HMWM, pentameric and tetrameric species (Fig. 1). After preliminary purification by silica chromatography, the mixture was subjected to preparative SEC. The HMWM and the nonamer overlapped substantially on the gravity-flow SEC columns. The use of THF instead of toluene as eluent or a decrease in the flow rate did not provide improved separation. Silica chromatography [CH2Cl2/hexanes (2∶1)] helped remove the HMWM to some extent (leading band on the column) but with significant loss of material (∼20%). In contrast, material losses were negligible with SEC. Accordingly, repetitive SEC was used in lieu of silica chromatography. Thus, a series of 7–9 SEC columns (for 20 mg of material) became necessary to obtain pure Zn8Fb as a red-purple solid (3.6 mg, 20% isolated yield). The purified sample of Zn8Fb gave a sharp peak in analytical SEC (Fig. 1C) and provided satisfactory absorption, 1H NMR and LD-MS data. Three side products were also isolated from SEC and analyzed by LD-MS: the intermediate pentamer Zn4Fb-I (<1%), and two phenyl-derivatized pentameric and tetrameric species of unidentified structure, Zn4Fb-Ph (<1%) and Zn4-Ph (2%). Metalation of Zn8Fb using Zn(OAc)2·2H2O afforded the all-Zn-nonamer Zn9.
 | ||
| Fig. 1 SEC traces for the synthesis of Zn8Fb using 1000, 500, and 100 Å columns in series. (A) Reaction mixture before the addition of Pd-catalyst. (B) Reaction mixture after 19 h. (C) Purified Zn8Fb. | ||
 | ||
| Scheme 3 Synthesis of nonameric arrays.50 | ||
(d) Pentamer Zn5-H. The synthesis of larger arrays was initiated by the preparation of a mono-ethynyl pentameric array. Thus, tetramer Zn4-H and I-Zn-TMS were subjected to the standard Pd-mediated coupling conditions (Scheme 4). After 4 h, the analytical SEC showed HMWM (9%), the desired pentamer along with a tetramer (80%), and monomeric species (10%) (data uncorrected for extinction coefficients). A combination of silica chromatography and SEC afforded pure Zn5-TMS in 66% yield. Analytical SEC of the purified sample showed a single sharp peak. The absorption, 1H NMR, LD-MS and HRMS data of Zn5-TMS were satisfactory. A putative phenyl-capped Zn4-porphyrin (Zn4-Ph, 10%) and octamer Zn8 (5%, vide infra) were also obtained as side products. Removal of the TMS group from Zn5-TMS with Bu4NF afforded Zn5-H in 95% yield.
 | ||
| Scheme 4 Synthesis of pentameric porphyrin building blocks.50 | ||
(e) 21-mer Zn20Fb. A convergent synthesis of Zn20Fb was performed by reaction of the Fb-porphyrin I4-Fb with four molar equivalents of pentamer Zn5-H under the standard Pd-coupling conditions (Scheme 5). After 3.5 h, analytical SEC indicated very slow reaction. Therefore, additional amounts of Pd2(dba)3 and AsPh3 were added. After 22 h, analytical SEC indicated incomplete consumption of monomer and pentamer but LD-MS analysis showed the formation of Zn20Fb (m/z obsd 16691.2, calcd 16693.0). Several products co-eluted on analytical SEC [HMWM, desired 21-mer, and other reaction products (16-mer and 11-mer resulting from incomplete coupling)] (Fig. 2). A further increase in the reaction time or the amount of Pd catalyst did not affect the yield of the array. This trend is in line with the multiple coupling reactions employed in the synthesis of Zn3Fb-TMS or Zn8Fb.
 | ||
| Fig. 2 SEC traces for the synthesis of Zn20Fb using a 103 Å column in series with a guard column. (A) Reaction mixture before the addition of Pd catalyst. (B) Reaction mixture after 3.5 h. (C) Reaction mixture after 22 h. (D) Purified Zn20Fb. | ||
 | ||
| Scheme 5 Synthesis of Zn20Fb and Zn21.50 | ||
The 21-mer could not be purified by the standard methods of column chromatography. After investigating a variety of separation methods (see Electronic Supplementary Information†), we turned to the use of semi-preparative SEC–HPLC columns. The optimum set of conditions for separation of the 21-mer on analytical SEC–HPLC was one 103 Å column or two 103 Å columns in series, a flow rate of 0.8 mL min−1, and THF as the mobile phase, but only 50–100 µg could be partially purified in each run. The use of a preparative HPLC–SEC (103 Å) column allowed larger sample injections (∼2 mg). A flow rate of 3.0 mL min−1 offered a good compromise between peak separation and run time [retention time (Zn20Fb) = 22.1 min], affording Zn20Fb as a broad peak with some tailing of porphyrins for over 15 min. Fractions (0.5–1.0 mL) were cut and impure fractions were rechromatographed. A total of 5–7 HPLC–SEC separations (for 5–8 mg of material) were carried out to yield pure Zn20Fb (1.9 mg, 19%). Analytical SEC of the purified sample gave a single sharp peak (Fig. 2D). Satisfactory absorption and MALDI-MS data were obtained but a well resolved 1H NMR spectrum was not obtained. Metalation of Zn20Fb with Zn(OAc)2·2H2O gave the all-zinc 21-mer Zn21.
In summary, the convergent synthetic approach affords diarylethyne-linked arrays in sufficient quantities for spectroscopic studies. The yields and ease of purification were satisfactory for the tetramer and pentamers (Zn3Fb-TMS, 61%; Zn5-TMS, 66%; asymmetric Zn4Fb, 81% yield) but the larger arrays were obtained in lower yield following extensive chromatography (Zn8Fb, 20%; Zn20Fb, 19% yield). We are currently working to develop refined approaches to obtain multiporphyrin arrays. It is noteworthy that a dendrimeric array similar in architecture to that of Zn20Fb, but incorporating 20 nickel porphyrins and one Fb-porphyrins with p-phenylene linkers, has been prepared via successive porphyrin-forming reactions.37
3. Use of SEC for the purification of multiporphyrin arrays
The successful purification of the larger multiporphyrin arrays proved challenging. We have employed SEC to purify and analyze multi-porphyrin arrays previously.2,22,23,47 However, with arrays comprised of more than five porphyrins, separation by SEC using the resins employed herein became extremely difficult. Arrays containing up to nine porphyrins were successfully purified by a combination of adsorption chromatography and repetitive SEC.The availability of a family of arrays of increasing size but otherwise similar architectures prompted the comparison of the chromatographic mobilities with analytical SEC columns of different pore size. The elution patterns of a family of seven arrays containing 1–5, 9 and 21 porphyrins on four different analytical SEC columns (100, 500, 103, 104 Å) are shown in Fig. 3. All arrays elute according to molecular size. The 500 Å or 103 Å column provides superior separation for all members of this set of arrays. The metalation state does not influence the retention time of the porphyrin array in SEC, as Fb-porphyrins and metalloporphyrins with the same structure eluted with nearly identical retention times.
 | ||
| Fig. 3 Analytical SEC elution profiles of porphyrin arrays using columns of different pore sizes. The labels “Zn4” and “Zn5” refer to Zn4-TMS and Zn5-TMS, respectively. | ||
4. LD-MS analysis of multiporphyrin arrays
In a prior study, we investigated the application of LD-MS and matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) to a collection of porphyrin monomers55 and a series of multiporphyrin arrays extending to a nonamer.56 Examination of the porphyrin monomers showed that the ionization process afforded intact molecule ion peaks (i.e., the cation radical rather than the protonated species). While porphyrin monomers and small arrays typically afford high quality spectra without a matrix (i.e., LD-MS), the study of the multiporphyrin arrays showed that the use of a matrix afforded higher quality spectra as the arrays increased in size (>5 porphyrins). Regardless of substrate, the soft ionization method of LD-MS or MALDI-MS preserves the metalation state of porphyrin arrays and allows successful analysis of arrays containing both Fb- and metallo-porphyrins.For the characterizations performed herein, intact, singly ionized molecules were observed for each of the arrays. Mass measurements agreed with the expected theoretical values within 0.3% accuracy. The matrices 4-hydroxy-α-cyano-cinnamic acid (4-HCCA) and ferulic acid (trans-4-hydroxy-3-methoxycinnamic acid), in saturated solutions of CHCl3 or toluene, were investigated for the analysis of the arrays comprised of 9 or 21 porphyrins. The best results (based on the signal intensity of the molecule ion peak under the same experimental conditions) were obtained using ferulic acid in toluene as matrix, followed closely by 4-HCAA in toluene. The spectrum with ferulic acid afforded a stronger molecule ion peak and less fragmentation (see Electronic Supplementary Information†).
All arrays containing four, five or nine porphyrins exhibited a similar fragmentation pattern with peaks separated by approximately 728 and 904 Da from the parent ion, corresponding to the loss of a peripheral trimesityl Zn-porphyrin and a trimesityl Zn-porphyrin containing a diphenylethyne linker, respectively, from the array. Some information was also obtained concerning the composition of the HMWM. The LD-MS data indicate that SEC fractions containing HMWM consisted of mixtures of materials in which one to 8–9 additional porphyrins are attached to a structure of mass corresponding to that of the desired array.
5. Solubility
All multiporphyrin arrays, with the exception of Zn4-H and Zn5-H, exhibit high solubility in toluene, THF, CHCl3 and CH2Cl2. The mesityl groups at the periphery of the arrays provide sufficient steric encumbrance of the porphyrin macrocycles to suppress cofacial aggregation. Moreover, the motion afforded by the flexibility (rotational freedom and bending)49 of the diphenylethyne linker permits good solubility relative to arrays containing more rigid linkages, such as molecular square arrays cyclo-Zn2Fb2U and cyclo-Zn4U.15 In contrast to their trimethylsilyl protected counterparts, Zn4-H and Zn5-H are only slightly soluble in halogenated solvents. Zn4-H shows relatively high solubility in toluene and THF, while Zn5-H is freely soluble only in THF. As one operational measure of solubility, we were able to obtain 1H NMR spectra of a number of the arrays at typical concentrations (∼1 mM) without noticeable aggregation.6. Photophysical properties
 | ||
| Fig. 4 Absorption spectra of Zn-porphyrin arrays in toluene at room temperature, showing the strong near-UV Soret B band(s) and the weaker visible Q bands. | ||
 | ||
| Fig. 5 Absorption spectra of the Zn-porphyrin arrays shown in Fig. 4, but focusing on the visible, Q-band region. | ||
| Compound | Soret (B) region | Visible (Q) region | ||||||
|---|---|---|---|---|---|---|---|---|
| λ max/nm | ε/M−1 cm−1 × 105 | λ max/nm | ε/M−1 cm−1 × 105 | λ max/nm | ε/M−1 cm−1 × 104 | λ max/nm | ε/M−1 cm−1 × 104 | |
| ZnTPP | 423 | 5.2 | 549 | 2.2 | 588 | 0.34 | ||
| Zn2 | 426 | 6.8 | 550 | 4.3 | 588 | 0.72 | ||
| Zn3 | 423 | 8.9 | 431 | 8.9 | 550 | 6.9 | 590 | 1.4 |
| Zn4-TMS | 424 | 14 | 431 | 14 | 550 | 11 | 591 | 2.2 |
| Zn5-TMS | 423 | 14 | 432 | 14 | 550 | 11 | 590 | 2.2 |
| Zn8Fb | 423 | 17 | 432 | 18 | 550 | 15 | 590 | 3.6 |
| Zn20Fb | 423 | 49 | 433 | 52 | 550 | 42 | 590 | 8.9 |
As the arrays progressively increase in size, some characteristics of the absorption spectra change appreciably while others do not. In the former category are the characteristics of the Soret (S0 → S2) absorption. Fig. 4 shows that the Soret band red shifts and splits (6–8 nm from Zn3 to Zn21) with increasing number of porphyrins in the array. These effects are well understood in terms of the pronounced exciton interactions between the large transition dipoles (reflected in ε ∼ 5 × 105 M−1 cm−1) of the Soret transitions of adjacent porphyrins, as we have described for star-shaped pentameric and smaller multiporphyrin arrays.19,20
On the other hand, the positions and widths of the Q bands (500–600 nm) remain virtually unchanged with increasing array size, except for a small red shift in the features (Fig. 5). The small spectral shift with increasing array size can be understood in terms of factors that include (1) the environmental effect of each Zn-porphyrin being surrounded by more porphyrins and less solvent than in smaller arrays, and (2) effects of relatively small interactions between the weak Q-transitions of adjacent porphyrins (for which the exciton splittings are expected to be less than the energy-widths of the bands58). It is also noteworthy that the molar decadic extinction coefficients (ε) of the Q bands scale approximately linearly with the number of Zn-porphyrins in an array. For example, for the Q(1,0) transition, Zn5-TMS exhibits ε = 110000 M−1 cm−1 which is 5 times that of ZnTPP (ε = 22000 M−1 cm−1)
(Table 1).
The observations on the Q bands reflect the fact that the linker-mediated inter-porphyrin interactions in the photophysically most relevant excited electronic state (S1) are relatively small. Furthermore, the results obtained here show that this is true even when considering the composite interactions that exist in an array with 21 porphyrins. The consequences include the following: (1) the inherent excited-state decay properties of the isolated porphyrin (microscopic rate constants for fluorescence, internal conversion, intersystem crossing) are retained for each porphyrin in the large arrays, and can be used as a benchmark for assessing additional processes that may occur in the arrays, such as excited-state energy transfer; (2) properties designed into the individual chromophores or smaller units will be retained (and need not be re-evaluated) as the architectures become increasingly larger and complex. We have previously noted these findings and expectations regarding the S1 excited state from detailed photophysical studies on smaller arrays,15,16,19,20 and find them to be manifest in the very large assemblies prepared and studied here. (Analogous conclusions regarding weak, but significant, inter-porphyrin electronic communication in the ground electronic state are drawn from the similarity in the redox properties of arrays with increasing size, as is described below.) It is also noteworthy that the increasing extinction coefficients of the Q-bands with increasing number of porphyrins in the array (Figures 4 and 5; Table 1) afford larger optical cross sections that enhance the light-harvesting function of the extended architectures relative to the smaller architectures.
The absorption spectra of the arrays containing a central Fb-porphyrin and either eight Zn-porphyrins (Zn8Fb) or 20 Zn-porphyrins (Zn20Fb) are shown in Fig. 6. These spectra are dominated by the spectral characteristics of the multiple Zn-porphyrins and are thus very similar to those of the all-Zn-porphyrin analogs Zn9 and Zn21 (Figures 4 and 5). The Soret band of the Fb-porphyrin is buried under the intense Soret absorption of the Zn-porphyrins; similarly, the Fb-porphyrin QY(0,0) and QX(1,0) bands at ∼550 and ∼590 nm are buried under the Zn-porphyrin Q(1,0) and Q(0,0) bands at these wavelengths. Close inspection of the visible-region spectra of Zn8Fb and Zn20Fb shows the presence (with the appropriate relative intensity) of the Fb-porphyrin QY(1,0) band at 515 nm, and particularly the QX(0,0) band at ∼650 nm that is well removed from any Zn-porphyrin features (see Electronic Supplementary Information†).
 | ||
| Fig. 6 Absorption spectra of Zn8Fb and Zn20Fb in toluene at room temperature. The insets show emission spectra in toluene using λex = 550 nm. | ||
The fluorescence spectra of the Zn8Fb and Zn20Fb arrays are dominated by emission from the central, single Fb-porphyrin component, even when the Zn-porphyrins are predominantly excited at 550 nm (Fig. 6, insets). These emission features are ascribable to the Fb-porphyrin QX(0,0) and QX(0,1) bands at ∼650 and ∼720 nm. The spectra of these large arrays also show a small amount of fluorescence from the large Zn-porphyrin pool, as is indicated by Q(0,0) Zn-porphyrin emission at ∼600 nm. Integration of the total emission (Zn- plus Fb-porphyrins from 565 to 800 nm) using predominant Zn-porphyrin excitation at 550 nm gives a total emission yield of 0.084 for Zn20Fb and 0.050 for Zn8Fb in toluene at room temperature. These values can be compared with the emission yields of Φf = 0.033 for ZnTPP and Φf = 0.11 for FbTPP.57,59 The observations that the total emission yields in the large arrays are less than that for FbTPP and that some residual Zn-porphyrin emission is observed both indicate that energy transfer to the central Fb-porphyrin is high, but not quantitative. However, it is difficult to draw quantitative conclusions of the energy-transfer efficiency from these emission studies alone (and the value for Zn8Fb seems anomalously low; vide infra). This is so because of (1) the technical difficulties associated with making detailed comparisons (which arise because of the large differences in relative absorption intensities in the large arrays and reference monomers), (2) the presence of emission from two types of porphyrins in the arrays, and (3) the effect on the apparent yields of only a minor Zn-porphyrin impurity in the large assemblies. Thus, estimates for the efficiency of energy transfer from the Zn-porphyrin light-harvesting components to the central Fb-porphyrin in Zn20Fb and ZnF8Fb are best made from the time-resolved absorption studies described below. These latter results are generally in good agreement with the emission data described above.
Fig. 7 shows representative absorption difference spectra for Zn8Fb in toluene at room temperature. The spectrum at 1 ps contains the features expected for Zn*, the most prominent of which is the bleaching of the Q(1,0) ground-state absorption band at ∼550 nm. Other Zn* features include the Q(2,0) bleaching at 510 nm, Q(0,0) bleaching and stimulated emission at ∼590 nm, and Q(0,1) stimulated emission at 650 nm. The Zn* features have largely decayed by 1.5 ns, and characteristics of Fb* have developed, as a result of energy funneling to the central porphyrin of the Zn8Fb array (Scheme 3). The most notable signatures of the overall Zn*Fb → ZnFb* energy-transfer process are the decay of Zn-porphyrin bleaching at 550 nm and growth of the Fb-porphyrin QY(1,0) bleaching at 515 nm (along with partial decay of the broad transient absorption extending from 450 to past 700 nm). Other Fb* features include QY(0,0) and QX(1,0) ground-state absorption bands at 550 and 590 nm, together with QX(0,0) bleaching and stimulated emission at 650 nm. Some of these features have similar positions to Zn* features, but have different intensity ratios as expected from the static optical spectra. In addition to the dominant contribution of Fb* to the 1.5 ns spectrum, there also appears to be some small residual Zn* contribution that most likely arises from monomeric pigments formed by minor decomposition.
 | ||
| Fig. 7 Time-resolved absorption spectra for Zn8Fb in toluene at room temperature, obtained using 130 fs excitation flashes at 550 nm. The inset shows a kinetic profile at 515 nm and a fit, giving an effective Zn* lifetime in the array of 105 ps (data before and during the flash and between 500 ps and 3.5 ns not shown for clarity). | ||
A representative time profile at 515 nm is shown in the inset to Fig. 7 (with data before and during the flash, and between 500 ps and 3.5 ns, not shown for clarity). The kinetic profiles at this and other wavelengths (e.g., 460 and 550 nm) are dominated by a component having a time constant of 105 ± 15 ps, obtained by averaging values from these regions on repeated samples. As is discussed below, this value represents the effective Zn* lifetime in the array, which decays essentially quantitatively via transfer of energy to the Fb-component. Inclusion of an additional, smaller amplitude component having a time constant of several nanoseconds gives the best fits, and this component is consistent with the presence of a small amount of monomeric Zn* resulting from minor decomposition. The presence of this kinetic component and the minor decomposition product does not affect the interpretation of spectral and kinetic data associated with the decay of Zn* or energy flow in the arrays. It is noteworthy that a fast component having a time constant of ∼10 ps is observed with increasing amplitude as the photon flux is progressively increased over that used for the measurements depicted in Fig. 7. This component is ascribed to Zn*Zn* → Zn*Zn excited-state annihilation involving multiple Zn* produced in a given array at high (but not low) excitation intensities.60 It is also noteworthy that the dominant 105 ps component (due to overall Zn*Fb → ZnFb* energy transfer) is unchanged within experimental error as the excitation intensity is varied.
Similar transient absorption studies were performed for the larger array Zn20Fb in toluene at room temperature. A time constant of 220 ± 40 ps is obtained from measurements at several wavelengths (e.g., 515 and 550 nm) on multiple samples using different excitation intensities. A fast component having a time constant of ∼7 ps is found at high, but not low, excitation intensities.
For comparison, the time constant for decay of the Zn bleaching and formation of the Fb bleaching for the ZnZnFb triad (Chart 2) is 90 ± 10 ps in toluene at room temperature (average of values in the two regions obtained here and previously20); this triad exhibits a fast component with a time constant of ∼3 ps at high excitation intensities. For the simpler ZnFbP dyad (Chart 1), the kinetic profiles in both the Zn- and Fb-porphyrin bleaching regions give a time constant of 45 ± 4 ps (average of values found here and previously20); the dyad shows no shorter-lived component at high excitation intensities because excitation of two Zn-porphyrins and consequent excited-state annihilation is not possible in this case. ZnFbP has the same methyl-group steric hindrance on the diarylethyne linker that is present between the Fb-porphyrin core and the adjacent Zn-porphyrins in Zn8Fb and Zn20Fb (Schemes 3 and 5). The related dyad ZnFbU, which has an unhindered linker,61 exhibits a shorter time constant of 24 ± 2 ps.20
The dominant time constant represents the average Zn* lifetime in each of the arrays; this time constant is independent of excitation intensity. When compared with the much longer ∼2.4 ns Zn* lifetime of monomeric Zn-porphyrins, it is also seen that these values are the same within error as the calculated average time constant for energy to arrive from the Zn-porphyrin pool to the Fb-porphyrin core.62 For each array, the energy-transfer time constant represents an average value that includes a range of overall transfer times to the core unit depending on which Zn-porphyrin is initially excited. In particular, the time depends on the distance of the initially excited Zn* to the core and thus, the number of transfer steps between Zn-porphyrins (which includes reversible back-and-forth transfers) before the final irreversible transfer to the central Fb-porphyrin. The data described above show that this overall transfer time to the core unit increases with increasing size of the array in the following order: ZnFbP (45 ps) < ZnZnFb (90 ps) < Zn8Fb (105 ps) < Zn20Fb (220 ps). Following established analyses,19,20,44,53 the lifetimes can be used to obtain (average) yields of energy transfer from the Zn-porphyrin(s) to the central Fb-porphyrin via the formula Φtrans = 1 − τA/τM, where τA is the time constant measured in the array and τM = 2.4 ns for the reference monomer. Thus, the overall energy-transfer efficiencies are ZnFbP (98%) < ZnZnFb (96%) < Zn8Fb (96%) < Zn20Fb (92%). The latter value is in reasonable agreement with the fluorescence quantum yield measured for this compound.
The trends and relative values of the overall transfer times for energy trapping at the core porphyrin are consistent with the structures of the various arrays. In particular, the minimal number of transfer steps between Zn-porphyrins (ignoring reversible back-and-forth hops) on a path directly to the central Fb-porphyrin will be, depending on which Zn-porphyrin is excited, as follows: either 2, 1, or zero for Zn20Fb; either 1 or 0 for Zn8Fb; either 1or 0 for ZnZnFb; and 0 for ZnFbP. Thus, it is reasonable that the smallest increment in transfer time between members of this series occurs between ZnZnFb (105 ps; Chart 2) and Zn8Fb (90 ps; Scheme 3); in both arrays the most peripheral Zn-porphyrin is only one Zn-porphyrin removed from the central Fb-porphyrin. The modestly longer arrival time at the core for Zn8Fb versus ZnZnFb can be explained in terms of the different topologies of the two arrays. In particular, for Zn8Fb more reversible transfers (hops) between Zn-porphyrins are possible owing to the branched architecture in each of the two Zn4 arms.
The above arguments also imply that the overall transfer time for Zn20Fb should be the longest among the arrays, as is observed. For this large array, energy-transfer processes can originate in the four most peripheral Zn-porphyrins, which are much farther from the Fb-porphyrin than are the two most peripheral Zn-porphyrins in Zn8Fb. Hence, more back-and-forth transfers can occur between Zn-porphyrins in Zn20Fb. Likewise, the fastest transfer times are expected for the ZnFbP (and ZnFbU) dyad because the sole Zn-porphyrin excited-state donor has no choice but to transfer energy to the adjacent Fb-porphyrin acceptor.
One of the primary objectives in preparing progressively larger arrays was to increase the light-harvesting ability of the architectures. The new Zn8Fb and Zn20Fb structures have achieved this goal. The enhanced light-harvesting ability with increasing size of the array is clearly reflected in the static optical spectra, which show the dramatically increasing extinction of the overlapping S0 → S1 absorption bands of the Zn-porphyrin light-harvesting chromophores relative to the single Fb-porphyrin energy trap. The light-harvesting ability of the arrays is also reflected in the static emission and time-resolved absorption characteristics, which reveal rapid energy transfer to the central unit. It is interesting to note that in going from the ZnZnFb triad to the Zn8Fb nonamer, there is a four-fold increase in the light-gathering ability (due to the increased number of Zn-porphyrins), but the average overall time for arrival of energy at the Fb-porphyrin core increases only modestly (from 90 to 105 ps). Again, these factors can be rationalized in terms of the structures of the triad and nonamer. However, more detailed considerations of the photodynamics in these two arrays (and Zn20Fb) must also take into account the fact that significant electronic communication between nonadjacent porphyrins can occur via a superexchange mediated process involving an intervening porphyrin.21 The communication between distant sites is expected to enhance the rates and efficiencies of energy transfer from the pool of light-harvesting pigments to the central porphyrin core.
7. Electrochemistry
We have previously investigated the electrochemical characteristics of dimeric (Zn2, ZnFbU, ZnFbP),4 trimeric (Zn3, ZnZnFb),4 and star-shaped pentameric (Zn4FbU,19 Zn5U,19 and others3) multiporphyrin arrays (Charts 1 and 2). Earlier bulk electrolysis studies of the star-shaped pentameric arrays showed that as many as eight electrons could be removed from the array to yield a stable, highly oxidized π-cation radical.3,19 Removal of additional electrons results in decomposition of the array. The fact that a large number of oxidizing equivalents can potentially be stored in multiporphyrin arrays prompted us to investigate hole storage in the largest array described herein, the 21-mer Zn20Fb.The voltammetric characteristics of Zn20Fb are similar to those we have measured for other multiporphyrin arrays.3,4,19 In particular, the 21-mer exhibits two reversible oxidation waves (E½(1) ∼0.5 V; E½(2) ∼0.9 V; versusAg/Ag+ in benzonitrile at room temperature; Fc/Fc+, E½ = 0.11 V). These waves correspond to the first and second oxidations of the porphyrin ring. The E½ values for the 20 weakly interacting Zn-porphyrins are very similar and cannot be resolved. In principle, the oxidation waves for the Fb-porphyrin should be resolvable (because the E½ values for this component are higher than those of the Zn- porphyrins3); however, the waves for the single Fb-porphyrin are completely masked by the waves from the large number of Zn-porphyrins. The redox potentials observed for the porphyrins in the 21-mer are essentially the same as those found previously in star-shaped pentameric and smaller arrays and in the isolated chromophores. This observation is indicative of relatively weak ground-state interactions between the constituent porphyrins in the 21-mer, as has been found previously for smaller arrays.3,4,19
Bulk electrolysis studies were carried out on Zn20Fb to investigate the hole-storage capabilities of this array. The progress of the bulk oxidation was monitored optically as shown in Fig. 8 which depicts the 500–1000 nm region of the absorption spectrum (the region in which porphyrin π-cation radicals exhibit the most characteristic spectral features3). As more electron equivalents are removed from the array, the broad, near-infrared optical signatures of a porphyrin π-cation become more pronounced. When 21 electron equivalents have been removed, the Q-band features characteristic of the neutral porphyrin have completely disappeared and only features of a porphyrin π-cation radical remain. These spectral characteristics indicate that the equivalent of one hole resides on each of the 21 porphyrins in the array. The (Zn20Fb)21+ species was relatively stable and exhibited no evidence of decomposition. The bulk-oxidized array was subsequently reduced with the recovery of ≥95% of the neutral material. The “super-charged” cation was also quite soluble and showed no evidence of precipitation. Attempts were not made to remove additional electrons from the array; however, the stability of (Zn20Fb)21+, along with our previous observation that up to eight holes can be stored in star-shaped pentameric arrays (Zn4FbU, Zn5U, others),3,19 suggest that even more holes could be reversibly stored in the 21-mer.
 | ||
| Fig. 8 Spectroelectrochemistry of (Zn20Fb)n+ in benzonitrile (n = 0, 2, 6, 12, 16, 19, and 21). The number on each spectrum represents the value of n. | ||
8 Conclusion
A convergent building block synthesis has been used to prepare a number of dendrimeric diarylethyne-linked porphyrin arrays, with the largest containing 21 chromophores. The light-harvesting ability of the arrays increases with the size of the array due to the increasing optical absorption cross-section afforded by the multiple Zn-porphyrins. The Zn-porphyrins funnel the excited-state electronic energy to the Fb-porphyrin at the core of the array. These same multiporphyrin arrays also serve as effective ground-state hole storage reservoirs, in that at least one electron can be removed from each chromophore in the assembly. The covalent diarylethyne linkages provide significant electronic communication between the chromophores but do not compromise characteristics that can be designed into building blocks, such as redox potentials and inherent photophysical properties of the lowest excited singlet state. Thus, this strategy complements approaches in which much stronger coupling between the units significantly modulates the properties of the units with increasing size and complexity of the overall array. From a synthetic standpoint, this building block approach enables facile synthesis of modestly sized arrays but the isolation of larger arrays in pure form requires extensive chromatography. The limitations encountered by the use of repetitive Pd-coupling reactions in this domain caused us (1) to develop refined conditions for Pd-coupling reactions with porphyrins,48 (2) to develop synthetic strategies that employ sequences of complementary chemistries in the construction of arrays,17,44 and (3) to investigate the use of non-covalent self-assembly of monomers and small covalently linked arrays to form the desired architecture.63 The good solubility, light-harvesting, and hole-storage properties of the dendrimeric arrays described herein, in conjunction with these new synthetic approaches, augurs well for the design and synthesis of larger multiporphyrin arrays of interest for a variety of applications.Acknowledgements
This research was supported by a grant from the Division of Chemical Sciences, Office of Basic Energy Sciences, Office of Energy Research, U. S. Department of Energy (to J. S. L.), and grants from the NIH (GM 36485 to D. H.; GM 39781 to D. F. B.). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the Facility was obtained from the North Carolina Biotechnology Center and the NSF. We thank Dr. Robert Donohoe for helpful discussions, Dr. Dongho Kim for assisting with some of the transient absorption measurements, and Prof. David C. Mauzerall for exploratory studies of multiphoton effects in the all-Zn-arrays.References
- A portion of this work was adapted from the Ph.D. thesis of Thomas E. Johnson, Department of Chemistry, Carnegie Mellon University, 1995.
- R. W. Wagner, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1996, 118, 11166 CrossRef CAS.
- J. Seth, V. Palaniappan, T. E. Johnson, S. Prathapan, J. S. Lindsey and D. F. Bocian, J. Am. Chem. Soc., 1994, 116, 10578 CrossRef CAS.
- J. Seth, V. Palaniappan, R. W. Wagner, T. E. Johnson, J. S. Lindsey and D. F. Bocian, J. Am. Chem. Soc., 1996, 118, 11194 CrossRef CAS.
- (a) D. P. Arnold, A. W. Johnson and M. Mahendran, J. Chem. Soc., Perkin Trans. 1, 1978, 366 RSC; (b) D. P. Arnold and L. J. Nitschinsk, Tetrahedron, 1992, 48, 8781 CrossRef CAS; (c) D. P. Arnold and L. J. Nitschinsk, Tetrahedron Lett., 1993, 34, 693 CrossRef CAS; (d) D. P. Arnold, D. A. James, C. H. L. Kennard and G. Smith, J. Chem. Soc., Chem. Commun., 1994, 2131 RSC.
- (a) S. Anderson, S. J. Martin and D. D. C. Bradley, Angew. Chem., Int. Ed. Engl., 1994, 33, 655 CrossRef; (b) H. L. Anderson, Inorg. Chem., 1994, 33, 972 CrossRef CAS.
- J. J. Gosper and M. Ali, J. Chem. Soc., Chem. Commun., 1994, 1707 RSC.
- (a) V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105 CrossRef CAS; (b) V. S.-Y. Lin and M. J. Therien, Chem. Eur. J., 1995, 1, 645 CrossRef CAS.
- (a) K. Susumu, T. Shimidzu, K. Tanaka and H. Segawa, Tetrahedron Lett., 1996, 46, 8399 CrossRef CAS; (b) A. Osuka and H. Shimidzu, Angew. Chem., Int. Ed. Engl., 1997, 36, 135 CrossRef CAS; (c) N. Yoshida, H. Shimidzu and A. Osuka, Chem. Lett., 1998, 55 CrossRef CAS; (d) T. Ogawa, Y. Nishimoto, N. Yoshida, N. Ono and A. Osuka, Chem. Commun., 1998, 337 RSC; (e) A. Nakano, A. Osuka, I. Yamazaki, T. Yamazaki and Y. Nishimura, Angew. Chem., Int. Ed., 1998, 37, 3023 CrossRef CAS; (f) T. Ogawa, Y. Nishimoto, N. Yoshida, N. Ono and A. Osuka, Angew. Chem., Int. Ed., 1999, 38, 176 CrossRef CAS; (g) N. Yoshida, N. Aratani and A. Osuka, Chem. Commun., 2000, 197 RSC; (h) N. Aratani, A. Osuka, Y. H. Kim, D. H. Jeong and D. Kim, Angew. Chem., Int. Ed., 2000, 39, 1458 CrossRef CAS.
- (a) C. Devadoss, P. Bharathi and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 9635 CrossRef CAS; (b) S. F. Swallen, R. Kopelman, J. S. Moore and C. Devadoss, J. Mol. Struct., 1999, 485–486, 585 CrossRef CAS; (c) S. F. Swallen, Z.-Y. Shi, W. Tan, Z. Xu, J. S. Moore and R. Kopelman, J. Lumin., 1998, 76–77, 193 CrossRef; (d) Y. Wakabayashi, M. Tokeshi, D.-L. Jiang, T. Aida and T. Kitamori, J. Lumin., 1999, 83–84, 313 CrossRef.
- M. A. Miller, R. K. Lammi, S. Prathapan, D. Holten and J. S. Lindsey, J. Org. Chem., 2000, 65, 6634 CrossRef.
- S. Prathapan, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1993, 115, 7519 CrossRef CAS.
- R. W. Wagner and J. S. Lindsey, J. Am. Chem. Soc., 1994, 116, 9759 CrossRef CAS.
- R. W. Wagner, J. S. Lindsey, J. Seth, V. Palaniappan and D. F. Bocian, J. Am. Chem. Soc., 1996, 118, 3996 CrossRef CAS.
- R. W. Wagner, J. Seth, S. I. Yang, D. Kim, D. F. Bocian, D. Holten and J. S. Lindsey, J. Org. Chem., 1998, 63, 5042 CrossRef CAS.
- J. Li, A. Ambroise, S. I. Yang, J. R. Diers, J. Seth, C. R. Wack, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1999, 121, 8927 CrossRef CAS.
- J. Li and J. S. Lindsey, J. Org. Chem., 1999, 64, 9101 CrossRef CAS.
- (a) D. Kuciauskas, P. A. Liddell, T. E. Johnson, S. J. Weghorn, J. S. Lindsey, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1999, 121, 8604 CrossRef CAS; (b) G. Kodis, P. A. Liddell, L. de la Garza, P. C. Clausen, J. S. Lindsey, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. B, in press Search PubMed.
- F. Li, S. Gentemann, W. A. Kalsbeck, J. Seth, J. S. Lindsey, D. Holten and D. F. Bocian, J. Mater. Chem., 1997, 7, 1245 RSC.
- J.-S. Hsiao, B. P. Krueger, R. W. Wagner, T. E. Johnson, J. K. Delaney, D. C. Mauzerall, G. R. Fleming, J. S. Lindsey, D. F. Bocian and R. J. Donohoe, J. Am. Chem. Soc., 1996, 118, 11181 CrossRef CAS.
- R. K. Lammi, A. Ambroise, T. Balasubramanian, R. W. Wagner, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 2000, 122, 7579 CrossRef CAS.
- N. Nishino, R. W. Wagner and J. S. Lindsey, J. Org. Chem., 1996, 61, 7534 CrossRef CAS.
- J. S. Lindsey, S. Prathapan, T. E. Johnson and R. W. Wagner, Tetrahedron, 1994, 50, 8941 CrossRef CAS.
- C. M. Drain, F. Nifiatis, A. Vasenko and J. D. Batteas, Angew. Chem., Int. Ed., 1998, 37, 2344 CrossRef CAS.
- H. Tamiaki, T. Miyatake, R. Tanikaga, A. R. Holzwarth and K. Schaffner, Angew. Chem., Int. Ed. Engl., 1996, 35, 772 CrossRef CAS.
- R. A. Haycock, A. Yartsev, U. Michelsen, V. Sundström and C. A. Hunter, Angew. Chem., Int. Ed., 2000, 39, 3616 CrossRef CAS.
- (a) J.-C. Chambron, V. Heitz and J.-P. Sauvage, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 6, pp. 1–42 Search PubMed; (b) J. Wojaczynski and L. Latos-Grazynski, Coord. Chem. Rev., 2000, 204, 113 CrossRef CAS; (c) T. Imamura and K. Fukushima, Coord. Chem. Rev., 2000, 198, 133 CrossRef CAS.
- (a) B. Wang, S.-W. Yang and W. E. Jones Jr, Chem. Mater., 1997, 9, 2031 CrossRef; (b) B. Jiang, S.-W. Yang, R. Niver and W. E. Jones Jr, Synth. Met., 1998, 94, 205 CrossRef CAS.
- B. Jiang, S.-W. Yang, D. C. Barbini and W. E. Jones Jr, Chem. Commun., 1998, 213 RSC.
- P. N. Taylor, J. Huuskonen, G. Rumbles, R. T. Aplin, E. Williams and H. L. Anderson, Chem. Commun., 1998, 909 RSC.
- (a) A. K. Burrell and D. L. Officer, SYNLETT, 1998, 1297 CrossRef CAS; (b) D. L. Officer, A. K. Burrell and D. C. W. Reid, Chem. Commun., 1996, 1657 RSC.
- (a) O. Mongin, C. Papamicaël, N. Hoyler and A. Gossauer, J. Org. Chem., 1998, 63, 5568 CrossRef CAS; (b) O. Mongin and A. Gossauer, Tetrahedron, 1997, 53, 6835 CrossRef CAS; (c) O. Mongin, N. Hoyler and A. Gossauer, Eur. J. Org. Chem., 2000, 1193 CrossRef CAS; (d) O. Mongin, A. Schuwey, M.-A. Vallot and A. Gossauer, Tetrahedron Lett., 1999, 40, 8347 CrossRef CAS.
- J. Wytko, V. Berl, M. McLaughlin, R. R. Tykwinski, M. Schreiber, F. Diederich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 1998, 81, 1964 CrossRef CAS.
- (a) S. Anderson, H. L. Anderson, A. Bashall, M. McPartlin and J. K. M. Sanders, Angew. Chem., Int. Ed. Engl., 1995, 34, 1096 CrossRef; (b) J. K. M. Sanders, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 3, pp. 347–368 Search PubMed.
- (a) H. Higuchi, M. Takeuchi and J. Ojima, Chem. Lett., 1996, 593 CAS; (b) R. Gauler and N. Risch, Tetrahedron Lett., 1997, 38, 223 CrossRef CAS; (c) R. Gauler and N. Risch, Eur. J. Org. Chem., 1998, 1193 CrossRef CAS.
- (a) O. Wennerström, H. Ericsson, I. Raston, S. Svensson and W. Pimlott, Tetrahedron Lett., 1989, 30, 1129 CrossRef; (b) D. Hammel, P. Erk, B. Schuler, J. Heinze and K. Mullen, Adv. Mater., 1992, 4, 737 CrossRef CAS; (c) A. Osuka, B.-L. Liu and K. Maruyama, Chem. Lett., 1993, 949 CAS; (d) A. Osuka, N. Tanabe, S. Nakajima and K. Maruyama, J. Chem. Soc., Perkin Trans. 2, 1996, 199 RSC.
- K. Sugiura, H. Tanaka, T. Matsumoto, T. Kawai and Y. Sakata, Chem. Lett., 1999, 1193 CrossRef CAS.
- T. Norsten and N. Branda, Chem. Commun., 1998, 1257 RSC.
- S. Hecht, T. Emrick and J. M. Fréchet, Chem. Commun., 2000, 313 RSC.
- K. Ichihara and Y. Naruta, Chem. Lett., 1995, 631 CAS.
- E. K. L. Yeow, K. P. Ghiggino, J. N. H. Reek, M. J. Crossley, A. W. Bosman, A. P. H. J. Schenning and E. W. Meijer, J. Phys. Chem. B, 2000, 104, 2596 CrossRef CAS.
- (a) N. Maruo, M. Uchiyama, T. Kato, T. Arai, H. Akisada and N. Nishino, Chem. Commun., 1999, 2057 RSC; (b) T. Kato, M. Uchiyama, N. Maruo, T. Arai and N. Nishino, Chem. Lett., 2000, 144 CrossRef CAS.
- (a) C. C. Mak, N. Bampos and J. K. M. Sanders, Chem. Commun., 1999, 1085 RSC; (b) C. C. Mak, N. Bampos and J. K. M. Sanders, Angew. Chem., Int. Ed., 1998, 37, 3020 CrossRef CAS.
- J. Li, J. R. Diers, J. Seth, S. I. Yang, D. F. Bocian, D. Holten and J. S. Lindsey, J. Org. Chem., 1999, 64, 9090 CrossRef CAS.
- D.-L. Jiang and T. Aida, J. Am. Chem. Soc., 1998, 120, 10895 CrossRef.
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467 CrossRef CAS; (b) H. A. Dieck and F. R. Heck, J. Organomet. Chem., 1975, 93, 259 CrossRef; (c) L. Cassar, J. Organomet. Chem., 1975, 93, 253 CrossRef CAS.
- R. W. Wagner, T. E. Johnson, F. Li and J. S. Lindsey, J. Org. Chem., 1995, 60, 5266 CrossRef CAS.
- R. W. Wagner, Y. Ciringh, P. C. Clausen and J. S. Lindsey, Chem. Mater., 1999, 11, 2974 CrossRef CAS.
- A. A. Bothner-By, J. Dadok, T. E. Johnson and J. S. Lindsey, J. Phys. Chem., 1996, 100, 17551 CrossRef CAS.
- The arrays are displayed for clarity with coplanar porphyrins in a 2-dimensional sheet-like conformation. However, free rotation about the ethyne and bending about the diphenylethyne linker49 enable a range of 3-dimensional conformations of the multiporphyrin arrays.
- A. Ambroise, R. W. Wagner, P. D. Rao, J. A. Riggs, P. Hascoat, J. R. Diers, J. Seth, R. K. Lammi, D. F. Bocian, D. Holten and J. S. Lindsey, Chem. Mater., 2001, 13, 1023 CrossRef CAS.
- The nomenclature for the dimers included a designation to identify the type of linker (U = unhindered; P = proximal hindered) joining the porphyrins. This linker terminology has not been included in the large arrays described herein.
- (a) J. P. Strachan, S. Gentemann, J. Seth, W. A. Kalsbeck, J. S. Lindsey, D. Holten and D. F. Bocian, J. Am. Chem. Soc., 1997, 119, 11191 CrossRef CAS; (b) J. P. Strachan, S. Gentemann, J. Seth, W. A. Kalsbeck, J. S. Lindsey, D. Holten and D. F. Bocian, Inorg. Chem., 1998, 37, 1191 CrossRef CAS; (c) S. I. Yang, J. Seth, T. Balasubramanian, D. Kim, J. S. Lindsey, D. Holten and D. F. Bocian, J. Am. Chem. Soc., 1999, 121, 4008 CrossRef CAS.
- C.-H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427 CrossRef CAS.
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, J. Porphyrins Phthalocyanines, 1999, 3, 283 CrossRef CAS.
- D. Fenyo, B. T. Chait, T. E. Johnson and J. S. Lindsey, J. Porphyrins Phthalocyanines, 1997, 1, 93 CrossRef CAS.
- M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. III, pp. 1–153 Search PubMed.
- M. Gouterman, D. Holten and E. Lieberman, Chem. Phys., 1977, 25, 139 CrossRef CAS.
- P. G. Seybold and M. Gouterman, J. Mol. Spectrosc., 1969, 31, 1 CrossRef CAS.
- With increasing number of light-harvesting units in an array (i.e., the number of Zn-porphyrins in our arrays) and/or increasing photon flux, there is the increasing possibility that more than one pigment may be excited simultaneously in a given array. The production of multiple excitations in a given array will have both spectral and kinetic consequences; such effects are observed for the Zn8Fb and Zn20Fb arrays at high but not at low excitation intensities. Even when present, these effects do not significantly impact the time constant determined for arrival of energy from the Zn pool to the Fb core, and may give an indication of the time scale for energy transfer between adjacent Zn-porphyrins in the arrays (See Electronic Supplementary Information†). The consequences of multiphoton effects on photophysical behavior are well known and have been explored extensively in photosynthetic antenna systems (Cf. D. Mauzerall, J. Phys. Chem., 1976, 80, 2306).
- The steric hindrance on the linker in ZnFbP (or analogous hindrance placed on the porphyrin) slows energy transfer relative to the unhindered linker in ZnFbU due to orbital overlap considerations.19,20,53 Likewise, the rate constants for energy hopping between Zn-porphyrins in the arrays will depend on the steric hindrance in the linker.
- Each of the measured time constants is rigorously the average Zn* lifetime of the Zn-porphyrin pool in the given array, which is ultimately dominated by energy transfer to the Fb core; this can be seen by comparison of each measured Zn* lifetime in an array (τA) with the Zn* lifetime of τM = 2.4 ns for the reference monomer. Following established analyses,19,20,44,53 the (average) transfer time from the Zn pool to the Fb core can be calculated using the expression τtrans = [(1/(τA)) − (1/(τM))]. (This equation assumes that the inherent rate constants for Zn* decay in the arrays are the same as in the isolated chromophore, which is a good approximation given the weak interporphyrin couplings and effects on the properties of the chromophores.) For Zn20Fb, the average energy-transfer time is τtrans = [(1/220 ps) − (1/2.4 ns)] = 240 ps, which is the same value within experimental error as the measured time constant of 220 ± 40 ps. The difference between the calculated transfer time and measured average Zn* lifetime decreases with decreasing lifetime and array size. Given these points, and the fact that the measured times already reflect average properties depending on which Zn-porphyrin is excited, we use the measured time constant as a good approximation of the average transfer time from the Zn-porphyrin pool to the Fb-porphyrin core in each array.
- A. Ambroise, J. Li, L. Yu and J. S. Lindsey, Org. Lett., 2000, 2, 2563 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: a description of multiphoton effects at high excitation intensities; the complete Experimental section including descriptions of the syntheses of the arrays; SEC data, 1H NMR spectra, and mass spectra for all new porphyrins and multiporphyrin arrays; a description of exploratory studies in the purification of Zn20Fb; data from a comparative study of analytical SEC conditions; representative mass spectral data under different conditions; and representative absorption and fluorescence spectra of the arrays. See http://www.rsc.org/suppdata/jm/b1/b105108n/ |
| ‡ Contribution from the Department of Chemistry, Carnegie Mellon University. Current address: Department of Chemistry, University of Georgia, Athens, GA 30602, USA. |
| This journal is © The Royal Society of Chemistry 2002 |