Synthesis of acicular hydrogoethite (α-FeOOH·xH2O; 0.1 < x < 0.22) particles using morphology controlling cationic additives and magnetic properties of maghemite derived from hydrogoethite
Chandran
Sudakar
,
G. Nagarajarao
Subbanna
and
T. R. Narayanan
Kutty
*
Materials Research Centre, Indian Institute of Science, Bangalore, India 560012. E-mail: kutty@mrc.iisc.ernet.in
First published on 28th November 2001
Abstract
A method for the preparation of acicular hydrogoethite (α-FeOOH·xH2O, 0.1 < x < 0.22) particles of 0.3–1 μm length has been optimized by air oxidation of Fe(II) hydroxide gel precipitated from aqueous (NH4)2Fe(SO4)2 solutions containing 0.005–0.02 atom% of cationic Pt, Pd or Rh additives as morphology controlling agents. Hydrogoethite particles are evolved from the amorphous ferrous hydroxide gel by heterogeneous nucleation and growth. Preferential adsorption of additives on certain crystallographic planes thereby retarding the growth in the perpendicular direction, allows the particles to acquire acicular shapes with high aspect ratios of 8–15. Synthetic hydrogoethite showed a mass loss of about 14% at ∼280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C, revealing the presence of strongly coordinated
water of hydration in the interior of the goethite crystallites. As evident from IR spectra, excess H2O molecules (0.1–0.22 per formula unit) are located in the strands of channels formed in between the double ribbons of FeO6 octahedra running parallel to the c-axis. Hydrogoethite particles constituted of multicrystallites are formed with Pt as additive, whereas single crystallite particles are obtained with Pd (or Rh). For both dehydroxylation as well as H2 reduction, a lower reaction temperature (∼220°C) was observed for the former (Pt treated) compared to the latter (Pd or Rh) (∼260°C). Acicular magnetite (Fe3O4) was prepared either by reducing hydrogoethite (magnetite route) or dehydroxylating hydrogoethite to hematite and then reducing it to magnetite (hematite–magnetite route). According to TEM studies, preferential
dehydroxylation of hydrogoethite along <010> leads to microporous hematite. Maghemite (γ-Fe2O3 − δ, 0 < δ < 0.25) was obtained by reoxidation of magnetite. The micropores are retained during the topotactic transformation to magnetite and finally to maghemite, whereas cylindrical mesopores are formed due to rearrangement of the oxygen sublattice from hexagonal to cubic close packing during the conversion of hydrogoethite to magnetite and then to maghemite. Accordingly, three different types of maghemite particles are realized: strongly oriented multicrystalline particles, single crystalline acicular particles with micropores or crystallites having mesopores. Higher values of saturation magnetization (σs = 74 emu g−1) and coercivity (Hc = 320 Oe)
are obtained for single crystalline mesoporous particles. In the other cases, the smaller size of particles and larger distribution of micropores decreases σs considerably (<60 emu g−1) due to relaxation effects of spins on the surface atoms as revealed by Mössbauer spectroscopy.
°C, revealing the presence of strongly coordinated
water of hydration in the interior of the goethite crystallites. As evident from IR spectra, excess H2O molecules (0.1–0.22 per formula unit) are located in the strands of channels formed in between the double ribbons of FeO6 octahedra running parallel to the c-axis. Hydrogoethite particles constituted of multicrystallites are formed with Pt as additive, whereas single crystallite particles are obtained with Pd (or Rh). For both dehydroxylation as well as H2 reduction, a lower reaction temperature (∼220°C) was observed for the former (Pt treated) compared to the latter (Pd or Rh) (∼260°C). Acicular magnetite (Fe3O4) was prepared either by reducing hydrogoethite (magnetite route) or dehydroxylating hydrogoethite to hematite and then reducing it to magnetite (hematite–magnetite route). According to TEM studies, preferential
dehydroxylation of hydrogoethite along <010> leads to microporous hematite. Maghemite (γ-Fe2O3 − δ, 0 < δ < 0.25) was obtained by reoxidation of magnetite. The micropores are retained during the topotactic transformation to magnetite and finally to maghemite, whereas cylindrical mesopores are formed due to rearrangement of the oxygen sublattice from hexagonal to cubic close packing during the conversion of hydrogoethite to magnetite and then to maghemite. Accordingly, three different types of maghemite particles are realized: strongly oriented multicrystalline particles, single crystalline acicular particles with micropores or crystallites having mesopores. Higher values of saturation magnetization (σs = 74 emu g−1) and coercivity (Hc = 320 Oe)
are obtained for single crystalline mesoporous particles. In the other cases, the smaller size of particles and larger distribution of micropores decreases σs considerably (<60 emu g−1) due to relaxation effects of spins on the surface atoms as revealed by Mössbauer spectroscopy.
1. Introduction
Preparation of fine particles having uniform size and specific shape is a fascinating and important area of research, crucial to applications in fields as diverse as catalysis, ceramics, magnetic recording media, etc.1,2 A wide range of classical routes to such materials have been developed over many years.3 Maghemite (γ-Fe2O3 − δ) particles for magnetic recording are commonly prepared by controlled oxidation of acicular magnetite (Fe3O4) particles.1,2,4 The only common, practical process for the preparation of acicular magnetite particle uses acicular goethite or lepidocrocite (α- or γ-FeOOH) as a starting material. Dehydration of α-FeOOH leads to particles of para(or heli)magnetic α-Fe2O3 (hematite), which are then reduced to yield Fe3O4. γ-FeOOH can be converted directly to γ-Fe2O3 − δ by dehydration, but this is difficult owing to the instability of γ-Fe2O3 − δ at elevated temperatures, as α-Fe2O3 can easily be the product of dehydration.5Different methods have been reported for the synthesis of acicular goethite particles. All the preparative methods can be broadly divided into two groups: (i) precipitation of ferrihydrates derived from Fe(NO3)3 or Fe2(SO4)3 solution by the slow addition of ammonia and transformation of these hydroxides to α-FeOOH by storage in KOH or NaOH solution,6–9 (ii) oxidative precipitation of FeCl2 or FeSO4 or (NH4)2Fe(SO4)2 in alkaline solution.10–14 In these methods, there are several factors, for the synthesis of α-FeOOH of characteristic shape and size, such as concentration of reagents, pH, temperature, time and the effect of various additives, which are cumbersome and time consuming to control. Also isomorphous replacement of some iron ions by trivalent (Al3+, Mn3+, V3+) and tetravalent (Ti4+, Sn4+) dopants influence the formation and structure of goethite.15,16 Synthetic and natural goethites display a variety of particle morphologies that are dependent on solution conditions during formation. Another factor that influences goethite morphology is the presence or absence of crystallo-specifically adsorbing ions in solution. It has been observed by many researchers17–20 that for the same conditions of hydrolysis and for a given concentration of Fe3+ ions, the nature of anions (Cl−, F−, ClO4−, NO3−, SO42−) affects the phase composition of hydrolytic products as well as the shape of the particles. Although several reports on the influence of sulfate21–23 and other anions on the formation of α-FeOOH are available, no simple method exists for the synthesis of α-FeOOH with acicular shape of high aspect ratio (length/breadth) and micrometer size. Here, we investigate the influence of cationic Pt, Pd and Rh additives in controlling the shape and size of α-FeOOH particles by anisotropic adsorption, i.e. preferential adsorption on certain crystallographic planes. The formation and properties of maghemite (γ-Fe2O3 − δ) prepared from the resulting needle shaped α-FeOOH with aspect ratio >10 have also been investigated.
2. Experimental
Acicular hydrogoethite particles were prepared by air oxidation of aqueous suspensions of ferrous ammonium sulfate (FAS) solution (0.1 M) containing 0.005–0.02 atom% of Pt4+, Pd2+ or Rh3+ cations (from Mn+(NH3)6Cln) at room temperature (25°C) under atmospheric pressure. Fe(OH)2·xH2O (80 < x < 120) aqueous suspensions were prepared by the addition of NH3 (aq) (1 M) to a solution of FAS. The amount of ammonia required for the precipitation was added in aliquots of 20–25 ml at intervals of 1.5 h followed by passing of air for oxidation. The aeration was continued for 24 h after complete precipitation of FAS, at a flow rate of 0.5–1 l min−1.
The pH was maintained between 4 and 7 throughout the reaction. Initially, the suspension was dark green and changed to lemon yellow or brown as the aeration continued and was accompanied by a decrease of pH from ∼7 to 4. The change in pH during oxidation was monitored by using a digital pH meter (Beckman, USA). Aliquots of samples were collected at different pH values from the mother-liquor to follow the phase and particle size evolution during the oxidation process. The precipitated hydrogoethite particles were washed with distilled water, dried in an oven (∼100°C) and then dispersed sonically in acetone or ethanol followed by filtration and drying. Experiments were also carried out under the same conditions, (i) without any additives and (ii) with organic additives such as sucrose, mannitol, polyvinylalcohol, etc., in the precipitation reaction medium. The best control of the acicular shape was obtained in presence of
Pt4+, Pd2+ or Rh3+ cations. Without any additives in the precipitation medium, matte aggregates of very small (<20 nm) acicular particles were obtained. With the organic additives, particles of mixed morphology are formed having acicular as well as spherical shapes. It is important to note that the amount of additives added to the solutions were low (0.005–0.02 atom%) so that they did not act as property modifying dopants but rather as growth regulating or morphology controlling agents. Further experiments using higher amounts of additives (up to 0.1 atom%) led to a decrease in aspect ratio of the particles. Samples prepared with 0.01 atom% of Pt4+, Pd2+ or Rh3+ ions in the precipitation medium showed maximum aspect ratio. The hydrogoethite particles so prepared using Pt and Pd (or Rh) as the cationic
additives are denoted G-I and G-II, respectively.
These hydrogoethite particles were simultaneously dehydroxylated and reduced (this process hereafter is referred to as the reduction process) under hydrogen at different temperatures ranging from 200 to 300°C for 2–4 h and then cooled in hydrogen to room temperature. The samples were black and the X-ray diffraction revealed the phase to be crystalline magnetite (Fe3O4) with complete conversion of FeOOH to Fe3O4 taking place. They possessed strong spontaneous magnetization when subjected to an applied magnetic field, which supported the formation of magnetite. The latter was oxidized in air at different temperatures between 150 and 250°C for 2 h, to obtain the maghemite phase (γ-Fe2O3 − δ).Samples were also dehydroxylated in air to hematite (α-Fe2O3) and reduced in hydrogen
to magnetite in two different steps. The samples were then oxidized to obtain maghemite. In this way, the difference in microstructural features such as pore shapes and distribution could be studied.
Details of the samples heat-treated at different temperatures in air or H2 and the corresponding labeling are given in Table 1. The steps of heat treatment, namely: dehydroxylation (D), reduction (R) and oxidation (O) are denoted in sequence separated by a slash (/), with the temperature of heat treatment denoted at the end. When dehydroxylation (D) and reduction (R) are carried out together, the step is denoted as DR (without slash). Thus, for example, G-IID/R/O260 represents the sample G-II dehydroxylated, reduced and oxidized in three different steps each being carried out at 260°C whereas G-IIDR/O260 represents the sample G-II dehydroxylated and reduced in one step under hydrogen atmosphere and then oxidized at 260°C.
| Sample identity | Heat treatment condition/°C | Phase identifieda | ||
|---|---|---|---|---|
| Dehydroxylation (D) | Reduction (R) | Oxidation (O) | ||
| a G = Goethite, γ = γ-Fe2O3 − δ, α = α-Fe2O3, M = magnetite, Fe = iron particles | ||||
| G-I (Pt 0.01 atom%) | – | – | – | G |
| G-II (Pd/Rh 0.01 atom%) | – | – | – | G |
| G-IDR/O220 | 220 | 220 | γ | |
| G-IDR260 | 260 | – | M, Fe | |
| G-IIDR/O220 | 220 | 220 | α, γ | |
| G-IIDR/O260 | 260 | 260 | γ | |
| G-IIDR300 | 300 | – | M, Fe | |
| G-IID/R/O260 | 260 | 260 | 260 | γ |
Phase identification of the powders was carried out by X-ray powder diffraction (Scintag/USA diffractometer using Cu-Kα radiation). Thermal analysis was performed on a simultaneous thermogravimetry/differential thermal analyzer (TG–DTA) from Polymer Laboratory, STA 1500, at a heating rate of 10°C min−1, in air or hydrogen atmosphere. Infrared absorption spectra were recorded on a Perkin-Elmer infrared spectrometer in the range 4000–300 cm−1 by dispersing the sample in anhydrous KBr pellets. Electron diffraction and microscopy were carried out with a JEOL, JEM 200CX, transmission electron microscope (TEM) for morphological and lattice imaging studies. Particle sizes and shapes were evaluated by the intercept method from the micrographs. To study the magnetization with respect to the applied field, a Vibrating Sample Magnetometer (VSM, Lakeshore, USA) was used. Mössbauer
spectra were recorded at constant acceleration in conjunction with a Nuclear Data Instruments ND60 multichannel analyser using a 57Co source in a rhodium matrix. The experimentally observed Mössbauer spectra were curve-fitted by a least-squares method by computer,24 assuming Lorentzian line shapes.
3. Results
The evolution of pH versus time during the preparation of sample G-II (Pd 0.01 atom%) by oxidation of the suspension with an initial ferrous salt concentration of 0.1 M is shown in Fig. 1. During the initial addition of an aliquot of ammonia (20 ml) the pH rises to 7.3 and then decreases as the air oxidation progresses with time. The pH decreases to 6.3 and remains constant for some time. This constancy of pH was then followed by a rapid decrease to pH ∼4, as the oxidation proceeds. When the pH reached around 4, the precipitate turned yellow–brown. With the addition of a fresh aliquot of ammonia, more ferrous hydroxide gel was precipitated and solid suspension acquired a mixed color between dark green and yellow brown. The duration of the constant pH interval decreased during the subsequent addition of aliquots of ammonia. Thus, two regions of decrease in pH are observed, one with a slower decrease (up to pH ∼ 6) and the second with a rapid decrease (to pH ∼ 4). The process of precipitation of Fe(OH)2·xH2O followed by air oxidation continued until all ferrous ions in the solution were completely precipitated and the gel was oxidized to generate goethite. The pH of the solution is around 7 after complete oxidation. It is observed during the precipitation experiments that the rapid increase of pH above 10 by the addition of excess ammonia leads to the formation of magnetite phase with a complete loss of acicular morphology. | ||
| Fig. 1 Change in pH as a function of time. Aliquots of NH3 (aq) (1 M) added at regular intervals are marked. | ||
3.1. Thermal analysis
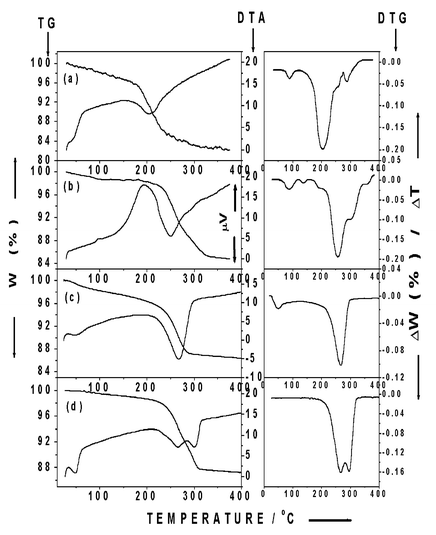
Thermal analyses of hydrogoethite samples were carried out both in air or hydrogen atmosphere. Fig. 2(a) and (b) show the simultaneous thermogravimetric (TG) and differential thermal analysis (DTA) curves. Sample G-I, consisting of multicrystallite acicular particles showed an endothermic peak at lower temperature (∼205°C) in H2 atmosphere while sample G-II, constituted of acicular particles of monocrystallite nature, showed a DTA (endothermic) peak around 250°C. The shift in temperature can be attributed to the difference in crystallite sizes. Thermal analysis of G-II carried out in hydrogen and in air is shown in Fig. 2(b) and (c), respectively. The endothermic peak (at 268°C) is broad and symmetric in air. The differential thermogravimetry (DTG) curve is sharp and the mass loss
is complete by around 300°C, whereas the endothermic peak in hydrogen atmosphere is broader (∼250°C) and the mass loss extends up to 350°C as observed from the DTG curve. It was found from X-ray diffraction that above 260°C the hydrogoethite phase converts to magnetite in an H2 atmosphere. Above 300°C, in H2 atmosphere, formation of iron through the reduction of Fe3O4 takes place, which accounts for the asymmetric and broader endotherm as well as the corresponding DTG curves. It was observed from TG/DTA curves of some of the samples (G-II) carried out in air that the DTA shows two resolved endothermic peaks as shown in Fig. 2(d) compared to that of a broad peak in Fig. 2(c). The first endotherm appears around 260°C and the second around 300°C. From the mass loss
through the broad range of temperature from 230 to 310°C together with the appearance of two resolved endotherms one may infer that initially there is a dehydration of hydrogoethite with some water loss leading to the first endotherm and then dehydroxylation to α-Fe2O3 with further mass loss, giving rise to the second endotherm. This, in turn, suggests that the endothermic peaks with corresponding mass losses arise from two different species (H2O and OH) rather than from a single species (OH group alone). In Fig. 2(c) such distinctly discernible endotherms were not observed and is indicative of the overlapping of the two endothermic steps, namely dehydration and dehydroxylation.
 | ||
| Fig. 2
TG–DTA curves of (a) G-I in H2, (b) G-II in H2, and (c), (d) G-II in air atmosphere. The corresponding DTG curves are given beside the TG-DTA curves. Curves (a)–(c) are at a heating rate of 10°C min−1; curve (d) is at 5°C min−1 (at a lower pressure of o.5 atm).
| ||
Mass loss measurements were also carried out under isothermal conditions by heating the samples for several hours at selected temperatures. The total mass loss (maximum) of synthetic hydrogoethite samples obtained in the present experiments was about 15%. After drying at 120°C for extended periods of ≈96 h, the residue shows a mass loss of about 14%
(maximum) at ∼280°C, which differs markedly from the theoretical value for stoichiometric FeOOH (10.13%). This indicates a compositional modification of goethite as FeO(OH)(H2O)x, where (H2O)x represents water of hydration present in the interiors of goethite crystallites. The compositions of hydrogoethite samples prepared with different additives, calculated from the percentage mass loss and Fe3+ estimated by volumetric analyses are given in Table 2.
The sample G-I has a lower hydroxy content compared to G-II. Natural hydrogeothite mineral samples with excess water contents are known in the literature.25
°C and γ-Fe2O3 − δ
| Sample identity | % of Fe (±0.2%) | wt% loss (up to 950°C) |
Composition of FeOOH calculated from wt% loss | |
|---|---|---|---|---|
| Goethite (atom% of additive) | G-I (Pt 0.01) | 61.14 | 12.87 | FeO(OH)(H2O)0.155 |
| G-II (Pd 0.01) | 60.53 | 13.77 | FeO(OH)(H2O)0.208 | |
| G-II (Rh 0.01) | 60.67 | 13.40 | FeO(OH)(H2O)0.186 | |
| Composition of γ-Fe2O3 − δ | ||||
| γ-Fe2O3 − δ | G-IDR/O220 | 70.43 | — | Fe2O2.93 |
| G-IIDR/O260 | 70.29 | — | Fe2O2.95 | |
| G-IID/R/O260 | 70.33 | — | Fe2O2.945 | |
3.2. Infrared spectra
Infrared spectra of the as-prepared samples are shown in Fig. 3(a) and (b). They are similar to the IR spectra of α-FeOOH26,27 but with the additional presence of absorption bands corresponding to water molecules. The appearance of a broad band at 3383 cm−1 due to O–H stretching vibration and an absorption band at 1636 cm−1 arising from the H–O–H bending vibration, are due to water present in the hydrogoethite. An absorption band at 3153 cm−1 is due to the O–H stretching mode of hydroxy groups. The sharp, distinct and intense pair of bands at 893 and 797 cm−1 are assigned to in-plane and out-of-plane Fe–O–H bending vibrations, respectively. The bands around 624 cm−1 and the doublet with maxima around 458 and 409 cm−1 are assigned to Fe–O stretching and Fe–OH symmetric and asymmetric stretching vibrations, respectively. The relative intensities of the O–H stretching bands at 3383 and 3153 cm−1 changes in the IR spectra on rehydration of the partially dehydroxylated G-II (in H2 at 220°C) as shown in Fig. 3(c). Also, the O–H stretching band at 3380 cm−1 and H–O–H bending mode at 1630 cm−1 of water show enhanced intensities as a result of rehydration of the sample. The O–H stretching and H–O–H bending absorption band positions indicate that the water molecules are coordinated rather than free.28 For free water, a much broader absorption peak is observed for the O–H stretching absorption band. The decrease in intensity of absorption bands corresponding to the bending modes of
Fe–OH groups together with the shift in the absorption band of Fe–O stretching from 624 to 553 cm−1 indicate partial conversion of FeOOH to FeO1 + x(OH)1 − 2x·yH2O and then to Fe2O3. The shoulder peak at 626 cm−1 can arise from the librational (breathing) mode of OH− groups. Thus, during dehydroxylation of FeOOH, in H2, rehydration becomes inevitable. Only after complete conversion either to Fe2O3
(by dehydroxylation in air) or to Fe3O4
(by dehydroxylation in H2), are anhydrous phases obtained, as shown in Fig. 3(d). The samples G-IDR/O220 and G-IIDR/O260 show absorption bands characteristic of γ-Fe2O3 − δ.When
the same samples are reduced in H2 at 300°C and reoxidized in air at 250°C very intense absorption bands below 600 cm−1 are observed.
 | ||
| Fig. 3
IR spectra of hydrogoethite samples: (a) G-I, (b) G-II, (c) partially dehydroxylated G-II (in H2 at 220°C) and (d) G-IIDR/O260.
| ||
The possible location of water molecules in the structure of goethite can be derived from the TG/DTA and IR spectral data. From the tetrahedral model,29 the typical environment of H2O (or OH−) consists of four neighbors arranged in a tetrahedron centered with O2− ions. H2O has an effective point charge +1/2 at two corners of the tetrahedron (with H+) and the other two corners have charge −1/2, whereas OH− has one corner +1/2 (with H+) and three corners −1/2. Corners of either sign are attracted electrostatically by oppositely charged corners of neighboring groups or ions of appropriate sign. The tetrahedral model suggests that H2O molecules and OH− groups play very similar roles in the structure and are equally able to form long or short hydrogen bonds. It is often difficult to distinguish whether a compound is a hydrate or a hydroxide. In the present case, the excess mass loss is due to H2O rather than OH− as confirmed from the IR spectra. A possible location for H2O in goethite is in the strands of the channels formed in between the double ribbons of FeO6 octahedra running parallel to the c-axis.29 Tetrahedron corners with charge −1/2 of H2O are hydrogen bonded to H from the edge shared OH groups of FeOOH, whereas the corners with +1/2 charge are hydrogen bonded to corner sharing oxygen ions. The random distribution of such water molecules in these channels accounts for the excess mass loss observed. The excess water ranges from 0.1 to 0.22 per formula unit of FeOOH, which is also the typical range observed in limonite or hydrogoethite minerals.25
3.3. XRD studies
Fig. 4(a) and (b) show X-ray diffraction (XRD) patterns of samples collected at pH ∼6 as well as ∼4, during the initial precipitation followed by air oxidation of sample G-II. Around pH 6, broad peaks of hydrogoethite together with that of an amorphous phase are observed. At pH = 4, where all the ferrous hydroxide gel was oxidized, peaks corresponding to hydrogoethite phase alone exist. The XRD patterns of completely crystallized hydrogoethite powders of G-I and G-II samples correspond to that of phase pure goethite (α-FeOOH) as shown in Fig. 4(c). However, the lattice parameters a = 4.659 Å, b = 9.989 Å and c = 3.0176 Å, show a slight increase along a and b compared the c axis.30 The samples were free from commonly found secondary phases during the oxidative precipitation of goethite, such as lepidocrocite (γ-FeOOH) or hematite (α-Fe2O3).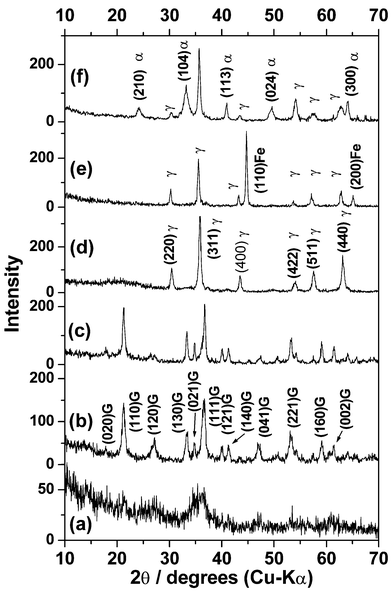 | ||
| Fig. 4 X-Ray diffraction patterns of sample collected (a) at pH ∼ 6 and (b) pH ∼ 4 during the initial precipitation followed by air oxidation of G-II, (c) hydrogoethite sample G-I, (d) G-IDR/O220, (e) G-IDR260 and (f) G-IIDR/O220. γ = γ-Fe2O3 − δ; Fe = metallic iron phase, G = hydrogoethite. | ||
The diffraction pattern obtained from sample G-I after reduction followed by reoxidation at 220°C, G-IDR/O220 (Fe = 70.43 wt%), corresponds to that of γ-Fe2O3 − δ
(Fig. 4(d)). When the sample was reduced at higher temperatures (>260°C) the XRD pattern (Fig. 4(e)) showed the presence of metallic Fe particles besides Fe3O4. Thus from sample G-I, phase-pure γ-Fe2O3 − δ was obtained when the heat treatment temperature was in the range 220–250°C. The XRD pattern of the samples obtained from G-IIDR/O220, showed a mixture of γ-Fe2O3 − δ and α-Fe2O3 phases (Fig. 4(f)). When the reduction temperature was higher (∼260°C), the reoxidized sample, G-IIDR/O260, showed phase pure γ-Fe2O3 − δ
(Fe = 70.29 wt%). With further increase of reduction temperature to 300°C, metallic Fe particles were formed along with γ-Fe2O3 − δ. Thus from G-II, phase-pure γ-Fe2O3 − δ was obtained when heat treatment conditions were within 260–280°C. From these observations it is clear that G-I sample transforms at lower heat treatment temperature to γ-Fe2O3 − δ as compared to G-II samples which is consistent with the results observed from TG–DTA curves.
Samples of γ-Fe2O3 − δ are also obtained from the corresponding hydrogoethite powders by initially dehydroxylating them in air at 220–280°C to hematite, α-Fe2O3, then reduction in H2, followed by re-oxidation in air.
3.4. TEM studies
The evolution of morphology in the course of synthesis of samples G-II (Pd or Rh ∼ 0.01 atom%) is shown in Fig. 5. Aggregates of small particles are observed at pH ∼7 as shown in Fig. 5(a). These particles were identified as amorphous hydrated ferrous hydroxide. Sample collected at pH ∼6 showed acicular particles of hydrogoethite crystallizing from the amorphous aggregate phase (Fig. 5(b)). As the oxidation proceeds, acicular particles grow and separate out from the amorphous mass as shown in Fig. 5(c). The conversion is not complete for these particles as amorphous aggregates are seen still to stick to the particles. At pH = 4.5 complete conversion into acicular hydrogoethite takes place. Most of these acicular particles at this stage have lower width as compared to the final product. The particles acquire a more tabular morphology when sampled at pH = 4 (Fig. 5(d)). Acicular hydrogoethite particles are evolved from the amorphous ferrous hydroxide by heterogeneous nucleation and growth involving an amorphous ferric oxyhydroxide intermediate phase rather than by dissolution of ferrous hydroxide followed by homogeneous nucleation from solution. | ||
| Fig. 5 Morphology evolution in the course of synthesis of sample G-II (Pd 0.01 atom%) during initial precipitation. Morphologies of samples collected at pH (a) 7, (b) 6, (c) 5.5 and (d) 4 are shown. | ||
Transmission electron micrographs (TEM) of acicular particles of hydrogoethite after complete precipitation are shown in Fig. 6. Sample G-I (Pt ∼ 0.01 atom%) shows highly oriented multicrystallites (Fig. 6(a)). The length of acicular particles is in the range 0.5–1 μm with an aspect ratio of 5–15. The shorter crystallites have an average length of ∼100 nm and aspect ratio of ∼5. Hydrogoethite particles, prepared using Pd or Rh as the morphology controlling cationic additives (G-II) are single crystallites with well-defined prism faces and truncated domal planes (Fig. 6(b)). The length of the particles is in the range 300–600 nm with aspect ratios in the range 7–15. Goethite particles prepared without any morphology controlling additives under the same conditions of precipitation showed (Fig. 6(c)) matte aggregates of small acicular particles of 10–20 nm length. The single crystal nature of sample G-II can be seen from the ED pattern shown in the inset of Fig. 6(d). The needle axis lies along [001] of the acicular hydrogoethite. The corresponding magnified image is shown in Fig. 6(d), wherein a few microchannels running along the needle axis can be seen. These may be the growth lineages (oriented dislocations formed during crystallite growth) between different regions of the same crystallites possibly due to dislocation bundles with apparent characteristics of ‘microchannels’.
 | ||
| Fig. 6 Electron micrograph of hydrogoethite samples: (a) G-I (Pt 0.01 atom%), (b) G-II (Pd 0.01 atom%) and (c) sample prepared without any additives. (d) Magnified image of a hydrogoethite particle (sample G-II) and the corresponding SAED pattern. | ||
As observed from TG–DTA and XRD data, sample G-I reduces to magnetite (Fe3O4) at 220°C and on re-oxidation at 220°C in air gives the maghemite phase. Fig. 7(a) shows the resulting morphology of maghemite particles. Though the acicular morphology persists, each particle by itself is made up of many crystallites. For G-II samples, the complete conversion to Fe3O4 takes place only around 265°C. Further oxidation at 250°C converts the magnetite to maghemite, γ-Fe2O3 − δ
(δ ≤ 0.2). The particles show near spherical pores of various radii in the range 5–15 nm (Fig. 7(b)). The size of these pores falls within the ranges of mesopores i.e. 2–50 nm. In some crystallites,
nearby pores join together to form cylindrical pores. However, the acicular particles do not disintegrate to smaller particles even in the presence of such mesopores. Maghemite obtained by the three-step process, i.e. dehydroxylation of α-FeOOH to α-Fe2O3, reduction to Fe3O4, followed by reoxidation in air, showed circular micropores (<2 nm) with diameter 0.8–1.8 nm (Fig. 7(c)), some of which align to form channels.
 | ||
| Fig. 7 Electron micrograph of γ-Fe2O3 − δ obtained from (a) G-I (G-IDR/O220) and (b) G-II (G-IIDR/O260), via the magnetite phase. (c) γ-Fe2O3 obtained from G-II through hematite–magnetite (G-IID/R/O260). A magnified image of one such particle is shown in the inset. | ||
Most of the γ-Fe2O3 − δ particles prepared from hydrogoethite by reduction followed by re-oxidation showed the <110> directions as the needle axis. The single crystal ED pattern of one such needle in Fig. 8(a), with the electron beam parallel to the [111] axis, is shown in the inset to Fig. 8(a). Also, occasionally <100> prevails as the direction of long axis in γ-Fe2O3 − δ as shown in Fig. 8(b). The ED pattern shows superlattice reflections along <100> directions, revealing the defect ordering in the unit cell of spinel to yield a larger cubic cell with a′ = 8.35 Å. A high-resolution electron micrograph (HREM) of the γ-Fe2O3 − δ particle is shown in Fig. 8(c). The variable thickness of the crystal, due to the presence of pores, causes the discontinuity in the lattice image. The fringes are found to orientate in the same direction, despite the discontinuity and variable thickness in the material, showing that the single crystal characteristics of the particle are preserved.
![(a)
γ-Fe2O3 − δ acicular particle obtained from G-II (G-IIDR/O260). The insert shows the corresponding ED pattern, (b) SAED pattern of another particle with the electron beam along [001] and (c) HREM of this particle.](/image/article/2002/JM/b104497b/b104497b-f8.gif) | ||
| Fig. 8 (a) γ-Fe2O3 − δ acicular particle obtained from G-II (G-IIDR/O260). The insert shows the corresponding ED pattern, (b) SAED pattern of another particle with the electron beam along [001] and (c) HREM of this particle. | ||
3.5. Magnetic measurements
The variation in magnetization with the applied field at room temperature for three different samples of γ-Fe2O3 − δ obtained from G-I and G-II are shown in Fig. 9. The magnetic properties as well as the pore shape and size of these particles are listed in Table 3. The saturation magnetization (σs) varies from 58.5 to 74.38 emu g−1 depending on the particle shape and size as well as the pore distribution. The corresponding remanent magnetization (Mr) ranges between 25.04 and 33.61 emu g−1. The coercivity of these samples varies from 280 to 320 Oe.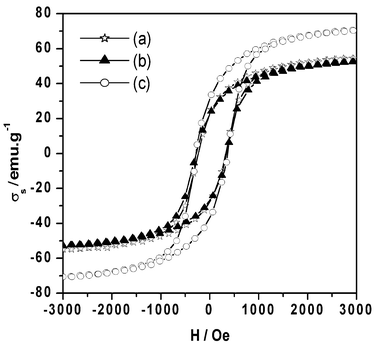 | ||
| Fig. 9 B–H curves of γ-Fe2O3 − δ obtained from (a) G-I (G-IDR/O220), (b) G-II (G-IID/R/O260), via hematite–magnetite and (c) G-II (G-IIDR/O260), via magnetite. | ||
| γ-Fe2O3 − δ | σ s (at 10 kOe) /emu g−1 | Coercivity/Oe | Squareness ratio | Nature of the particle observed |
|---|---|---|---|---|
| GI-DR/O220 | 58.5 | 280.1 | 0.42 | Acicular particles with multicrystallites |
| G-IIDR/O260 | 74.38 | 319.3 | 0.45 | Acicular single crystals (with cylindrical pores of 5–15 nm) |
| G-IID/R/O260 | 56.5 | 300.1 | 0.43 | Acicular particles having circular pores of 0.8–1.8 nm |
The difference in saturation magnetization of γ-Fe2O3 − δ obtained from G-I and G-II can be understood from the microstructural features of the particles. G-I consists of acicular particles with multicrystallites constituting each grain whereas G-II is made up of particles with monocrystalline characteristics. The individual crystallite sizes of the γ-Fe2O3 − δ particles, G-IDR/O220, are small (<50 nm) so that they are superparamganetic in nature. Mallinson31 has reported that for γ-Fe2O3 − δ, relaxation effects in zero field will appear for spherical particles with diameters <70 nm and for acicular particles with an aspect ratio of 5 and length <75 nm. However, the Mössbauer spectra obtained for this sample (Fig. 10) at room temperature shows the absence of an asymmetric doublet. This indicates that the crystallites that constitute the needle-shaped particle themselves are strongly oriented. However, the large surface area of the highly oriented small particles lowers σs due to relaxation effects of spins of magnetic atoms lying on the surface. For γ-Fe2O3 − δ with circular micropores of 0.8–1.8 nm, σs and Hc (56 emu g−1 and 300 Oe) are lower than samples containing cylindrical mesopores of size 5–15 nm. The decrease in coercivity arises from the increase in void volume within the particles. It is very clear from the TEM micrographs that the micropore distribution in γ-Fe2O3 − δ is more frequent and uniformly distributed than the mesopores and hence the increase of void volume in microporous γ-Fe2O3 − δ. It is well known that any type of defect contributes to lowering of the energy barrier for irreversible magnetization and demagnetization.32 This material discontinuity due to larger distribution of circular micropores also reduces σs values to considerably lower levels than the σs of γ-Fe2O3 − δ with cylindrical mesopores.
 | ||
| Fig. 10 Mössbauer spectra of γ-Fe2O3 − δ at room temperature: (a) G-I (G-IDR/O220) and (b) G-II (G-IIDR/O260). | ||
3.6 Mössbauer spectra
The Mössbauer spectra of γ-Fe2O3 − δ derived from G-I and G-II with magnetite as the intermediate are shown in Fig. 10. The computed Mössbauer parameters for these samples are given in Table 4. The values of the isomer shift (IS) show that the iron is in trivalent state in both the samples. Nearly zero values of quadrupole splitting (QS) for the sextets, a typical characteristic of spinel ferrites, indicate the presence of γ-Fe2O3 − δ with the defect spinel ferrite structure.33 The sextets observed indicate that the fields at the octahedral and tetrahedral sites are about the same, to within a few percent. However the slight asymmetry in spectral lines (Fig. 10(a)) for γ-Fe2O3 − δ (derived from G-I) infers the presence of two hyperfine interactions. Accordingly the effective magnetic field calculated from the fitted spectral data, assuming lines to be Lorentzian in shape, are given in Table 4. The Heff for both samples from sextet-I showed the hyperfine interaction of bulk ferromagnetic γ-Fe2O3 − δ, corresponding to Fe3+ ions situated in the cores of the nanoparticles.34Heff from sextet-II of the spectra showed a decrease in effective hyperfine interaction. The smaller Heff value as compared to bulk γ-Fe2O3 − δ is due to the small particle size and can be associated with Fe3+ ions at or near the surface. Further, the area under sextet-I (56.2%) and sextet-II (43.8%) are almost equal in γ-Fe2O3 − δ derived from G-I whereas for γ-Fe2O3 − δ derived from G-II, sextet-II has a much smaller area (21.7%) indicating that the effective contribution from the surface atoms is very small. It is also clearly evident from TEM studies that γ-Fe2O3 − δ derived from G-II are acicular single crystallite particles whereas γ-Fe2O3 − δ derived from G-I are multicrystallite particles of smaller size (<50 nm). γ-Fe2O3 − δ derived from G-II with hematite–magnetite formation in the intermediate steps, G-IID/R/O260, also a showed contribution from the surface Fe3+ ions with Heff = 491 kOe and area under the corresponding sextet-II equal to 40.8%. However, the linewidth for sextet-II is considerably narrower for γ-Fe2O3 − δ derived from G-II samples compared to G-I.| γ-Fe2O3 − δ sample origin | Sextet-I | Sextet-II | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IS | QS | LW | H eff | Area | IS | QS | LW | H eff | Area | |
| a IS = Isomer shift (mm s−1), QS = quadrupole splitting (mm s−1), LW = line width (mm s−1), Heff = hyperfine magnetic field (kOe), Area = area under the sextet (%). | ||||||||||
| G-IDR/O220 | 0.363 | -0.10 | 0.46 | 509 | 41.9 | 0.371 | 0.00 | 1.06 | 481 | 58.1 |
| G-IIDR/O260 | 0.337 | 0.01 | 0.54 | 503 | 78.3 | 0.305 | -0.07 | 0.59 | 492 | 21.7 |
| G-IID/R/O260 | 0.323 | -0.01 | 0.47 | 506 | 59.2 | 0.322 | -0.00 | 0.84 | 491 | 40.8 |
4. Discussion
Acicular hydrogoethite particles are prepared from aqueous suspensions of ferrous hydroxide in the presence of growth regulating cationic Pt, Pd and Rh additives. The process of formation of α-FeOOH from Fe(OH)2·xH2O by air oxidation occurs viaeqn. (1):| Fe(OH)2·xH2O → FeOx(OH)3 − 2x·yH2O + (2x − 1)H+ | (1) |
| Fe2+ + OH− → Fe3+ + O2− + H+ | (2) |

The goethite samples contained excess water molecules, located in the strands of channel formed in between the double ribbons of FeO6 octahedra running parallel to the c-axis. During the growth and recrystallization from FeOx(OH)3 − 2x·yH2O some excess water is residually retained in FeOOH, leading to hydrogoethite, FeO(OH)(H2O)x, with x ∼0.1–0.22. The excess water present in the goethite is evident from the IR spectra. The crystallite size and oriented aggregation of the particles, however, may differ depending on the additive used. Thus for hydrogoethite sample G-I, each acicular particle consists of highly oriented small crystallites of multicrystalline needles, whereas sample G-II is comprised of single crystallite acicular particles. G-I samples show lower reaction temperature (∼220°C),
compared to G-II samples (260–270°C) both in dehydroxylation as well as in H2 reduction. The lower conversion temperature of G-I is due to the higher disorder arising from the multicrystallinity of individual particles.
γ-Fe2O3 − δ obtained from G-I, G-IDR/O220, showed acicular particles containing microcrystallites, the crystallites being highly oriented within any given particle. γ-Fe2O3 − δ obtained from G-II showed single crystalline particles with pores. The pore size and distribution differed by way of γ-Fe2O3 − δ preparation. When γ-Fe2O3 − δ is obtained via the hematite–magnetite route, circular micropores (of width 0.8–1.8 nm) were observed. Through the magnetite route (carrying out dehydroxylation and reduction together), mesopores of cylindrical nature are observed. During the hydrogoethite–hematite transformation, preferential dehydration along the <010> direction leads to the formation of a microporous texture on the particle surface.40 This pore structure is retained during the topotactic transformation from hematite to magnetite and finally to maghemite, under controlled heat-treatment conditions, with only minor change in the texture. When the dehydroxylation and reduction are carried out together to obtain maghemite directly from hydrogoethite, the oxygen sublattice rearranges from hexagonal to cubic close packing by way of changing the stacking sequence or by relative rotation of the adjoining octahedra. This leads to a gathering of pores because of increased mobility of iron ions and counter-mobility of vacancies. Hence mesopores are formed during the direct transition from hydrogoethite to magnetite. Under controlled heat-treatment conditions the acicular morphology is still preserved without disintegration of the particle to finer crystallites.
Magnetic measurements on the particles obtained by three different routes showed differences in coercivity and saturation magnetization. Higher coercivity (320 Oe) is observed for particles with cylindrical mesopores. The particles with micropores have a coercivity of 300 Oe and particles with multicrystalline nature showed a coercivity of 280 Oe. The saturation magnetization was highest for the particles with cylindrical mesopores (74.38 emu g−1), while the saturation magnetization was observed to be nearly the same for highly oriented multicrystalline particles and the particles with circular micropores (58.5 and 56.5 emu g−1, respectively). A larger distribution of micropores and smaller size of particles decreases σs considerably due to relaxation effects of spins on the surface atoms.
5. Conclusions
There are considerable differences among the available reports on the process of formation of acicular goethite particles and the magnetic properties of maghemite derived from goethite.2 The precipitation processes, however, are too complicated to be carried out in a controlled fashion to realize desired particle sizes and shapes. The magnetic properties are often difficult to optimize to the desired level of magnetocrystalline anisotropy for use in memory devices. Thus investigations are still continuing on simple methods of formation of acicular goethite particles and of maghemite with optimum magnetic properties. We have reported here the synthesis of uniform acicular hydrogoethite particles by air oxidation of aqueous suspensions of ferrous salts in the presence of morphology controlling cationic additives (Pt, Pd or Rh). With Pd (or Rh) the hydrogoethite particles are single crystallites with aspect ratio >10 and are suitable precursors for obtaining high-density magnetic storage materials. The magnetic properties of the maghemite particles obtained from hydrogoethite are as good as commercially available γ-Fe2O3 − δ. The present results are a signicant step forward in a comprehensive study involving the preparation aspects and precipitation processes to achieve acicular hydrogoethite, the complexity that prevails in microstructures and of topotactic transformations in iron oxyhydroxides to iron oxides with respect to heat-treatment conditions and further optimizing the magnetic properties of maghemite.References
- G. Bate, in Magnetic Oxides, Part 2, , ed. D. J. Craik, Wiley, New York, 1975, p 689 Search PubMed.
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory, Wiley-VCH, Weinheim, 2nd edn., 2000 Search PubMed.
- E. Matijevic, Chem. Mater., 1993, 5, 412 CrossRef CAS.
-
M. Camras, US Pat., 2694656, 1954 Search PubMed.
- M. P. Sharrock, IEEE Trans.Magn., 1989, 25(6), 4374 CrossRef CAS.
- U. Schwertmann, Thermochim. Acta., 1984, 78, 39 CrossRef CAS.
- D. G. Schulze, Clays Clay Miner., 1984, 32, 36 Search PubMed.
- D. G. Schulze and U. Schwertmann, Clay Miner., 1984, 19, 521 Search PubMed.
- J. Torrent, V. Barron and U. Schwertmann, Soil. Sci. Soc. Am. J., 1987, 51, 78 Search PubMed.
- M. Kiyama, Bull. Chem. Soc. Jpn., 1974, 47, 1646 CAS.
- C. Domingo, R. Rodriguez-Clemente and M. A. Biesa, Solid State Ionics, 1993, 59, 187 Search PubMed.
- D. Concepcion, R. Rapael and B. Miguel, J. Colloid Interface Sci., 1994, 165, 244 CrossRef.
- S. Music, S. Popovic and M. Gotic, J. Mater.Sci., 1990, 25, 3186 CrossRef CAS.
- K. Kandori, M. Fukuoka and T. Ishikawa, J.Mater. Sci., 1991, 26, 3313 CAS.
- F. J. Berry, Ö. Helgason, A. Bohórquez, J. F. Marco, J. Mc Manus, E. A. Moore, S. Mørup and P. G. Wynn, J. Mater. Chem., 2000, 10, 1643 RSC.
- T. Ishikawa, H. Yamashita, A. Yasukawa, K. Kandori, T. Nakayama and Fumio Yuse, J. Mater. Chem., 2000, 10, 543 RSC.
- C. Domingo, R. Rodriguez-Clemente and M. A. Blesa, J. Colloid Interface Sci., 1994, 165, 244 CrossRef.
- E. Matijevic and P. Schiener, J.Colloid Interface Sci., 1978, 63, 509 CAS.
- J. Dousma, T. J. Van Den Hoven and P. L. De Bruyn, J. Inorg. Nucl. Chem., 1978, 40, 1089 Search PubMed.
- J. Dousma, D. Den Ohelander and P. L. De Bruyn, J. Inorg. Nucl. Chem., 1979, 41, 1565 Search PubMed.
- S. Music, A. Vertes, G. W. Simmons, I. Czaknagy and H. Jr. Leidheiser, J.Colloid Interface Sci., 1982, 85, 256 CrossRef CAS.
- S. Music, Z. Orehover, S. Popovic and I. Czako-Nagy, J. Mater. Sci., 1994, 29, 1991 CAS.
- K. M. Parida and J. Das, J. Colloid Interface Sci., 1996, 178, 586 CrossRef CAS.
- E. Von Meerwall, Comput. Phys. Commun., 1975, 9, 117 CrossRef.
- A. S. Marfunin, Spectroscopy, Luminescence and Radiation Centers in Minerals, Springer-Verlag, Berlin, Heidelberg, 1979, p. 33 (and references therein) Search PubMed.
- L. Verdonck, S. Hoste, F. F. Roelant and G. P. Vander kelen, J. Mol. Struct., 1982, 79, 273 CrossRef CAS.
- B. Weckler and H. D. Lutz, Eur. J. Solid State Inorg.Chem., 1998, 35, 531 CrossRef CAS.
- K. Nakamoto, Infrared Specta of Inorganic and Coordination Compounds, John Wiley & Sons, Inc., London, 1963 Search PubMed.
- H. D. Megaw, Crystal Structures: A Working Approach, W. B. Saunders Company, London, 1973 Search PubMed.
- A. F. Fualtieri and P. Venturelli, Am. Mineral., 1999, 84, 890 Search PubMed.
- T. C. Mallinson, in Magnetic Properties of Materials, ed. J. Smit, McGraw Hill, New York, 1971, ch. 7 Search PubMed.
- S. J. Andress, A. Benedetti, A.R. Corradi and G. Fagherazzi, IEEE Trans Magn., 1986, 22(5), 1341 CrossRef.
- M. N. Greenwood and T. G. Gibbs, Mössbauer Spectroscopy, Chapman & Hall, London, 1976 Search PubMed.
- J. Danon, Chemical Applications of Mössbauer Spectroscopy, ed. V. I. Goldanski, and R. H. Herber, Academic Press, New York and London, 1968 Search PubMed.
- T. R. N. Kutty, unpublished work.
- A. Manceau and L. Charlet, J. Colloid Interface Sci., 1994, 168, 87 CrossRef CAS.
- L. Spadini, A. Manceau, P. W. Schindler and L. Charlet, J. Colloid Interface Sci., 1994, 168, 73 CrossRef CAS.
- J. D. Ostergren, G. P. Trainor, J. R. Bargar, G. E. Brown Jr. and G. A. Parks, J. Colloid Interface Sci., 2000, 225, 466 CrossRef CAS.
- J. D. Ostergren, G. E. Brown Jr., G. A. Parks and P. Persson, J. Colloid Interface Sci., 2000, 225, 483 CrossRef CAS.
- H. Naono and R. Fujiwara, J. Colloid Interface Sci., 1980, 73, 406 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |