Determination of trace metals in sea-water by electrothermal atomic absorption spectrometry following solid-phase extraction: quantification and reduction of residual matrix effects
Marco Grotti*, Maria Luisa Abelmoschi, Francesco Soggia and Roberto Frache
Department of Chemistry and Industrial Chemistry, University of Genoa, via Dodecaneso 31, I-16146 Genova, Italy. E-mail: grotti@chimica.unige.it
First published on 23rd November 2001
Abstract
The use of an iminodiacetic resin for trace metal preconcentration from sea-water does not allow a complete removal of the matrix, since significant amounts of alkali and alkaline earth elements are retained by the resin and co-elute with the analytes. The interfering effects due to sodium, potassium, calcium and magnesium on the electrothermal atomization of cadmium, lead, copper, iron and manganese were studied in a multivariate way. The adopted method allowed both an accurate quantification of the signal variations due to the matrix elements and a prediction of the analytical error in the determination by electrothermal atomic absorption spectrometry. In order to reduce the interfering effects, two different approaches were considered: chemical modification and matrix separation by pre-elution with ammonium acetate. A significant decrease of the interfering effects was observed for copper, lead and cadmium, after optimization of the thermal programs and proper choice of the chemical modifier. Differently, strong signal variations caused by the matrix were observed for iron and manganese, for each condition. Matrix separation by selective elution with ammonium acetate was more effective, allowing the complete suppression of matrix effects for all the considered elements. The results were confirmed by the analyses of the sea-water reference materials CASS-3 and NASS-5.
Introduction
There is a great interest in the determination of trace elements in sea-water, including routine monitoring of marine environmental pollution, investigations on the chemistry of marine systems and studies on the biogeochemical cycles of the elements.Although electrothermal atomic absorption spectrometry (ETAAS) is a well-established technique for trace metal determination in aqueous solutions, the direct analysis of sea-water is troublesome, due to the extremely low analytical concentrations and strong interference arising from the matrix.
As a consequence, several procedures have been proposed for preconcentration of the analytes and separating from the interfering matrix prior to measurement by ETAAS. Notable approaches have made use of solution-phase chelation followed by adsorption onto reversed-phase C18 substrates1–5 or PTFE knotted reactors,6–8 retention on chelating resins,9–17 precipitation18–22 and electrodeposition.23
Flow injection as a micro-sample introduction system offers some advantages over manual batch-type procedures, such as fully automated sample treatment and low contamination. On the other hand, the batch equilibration procedures do not require the use of sophisticated instrumentation and allow the preconcentration step to be carried out directly on board of an oceanographic ship, preventing the risks related to the conservation of samples.
In any case, the preconcentration procedure using a chelating resin, though able to eliminate most of the marine salts, is not completely selective for trace metals. Hence, a fraction of the major elements (mainly calcium and magnesium) is usually retained by the resin, co-elutes with the analytes and is introduced into the atomizer.
In this work, the interferences due to sodium, calcium, potassium and magnesium, still present after the preconcentration step and affecting the electrothermal atomization of cadmium, lead, copper, iron and manganese, were evaluated in a multivariate way. In order to reduce the observed matrix effects, two methods were compared: chemical modification and matrix separation by pre-elution with ammonium acetate.
Experimental
Apparatus
ETAAS measurements were carried out using a Varian (Springvale, Australia) SpectrAA 300 atomic absorption spectrometer equipped with Zeeman-effect background correction and a GTA 96 graphite atomizer. Samples and modifier solutions were delivered to the furnace using a Varian PSC 56 programmable sample changer, after storing in acid washed polypropylene cups. Pyrolitic graphite-coated tubes with or without pyrolytic platforms were used. The temperature programs, reported in Tables 1 and 2, were optimised in the presence of the matrix in order to get the highest absorbance value and maximum pyrolysis temperature without loss of analyte. Integrated absorbance was measured exclusively. Other operating parameters are listed in Table 3.| Step | ||||||
|---|---|---|---|---|---|---|
| Parameter | 1 | 2 | 3 | 4 | 5 | 6 |
a Pyrolis temperature (see Table 2).b Atomization temperature (see Table 2).c Cleaning temperature, set 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C higher than the corresponding atomization temperature. °C higher than the corresponding atomization temperature. | ||||||
| Temperature/°C | 50 | 95 | 120 | TPyra | TAtomb | TCleanc |
| Ramp time/s | 5 | 60 | 10 | 10 | 0 | 2 |
| Hold time/s | 0 | 0 | 0 | 10 | 2 | 0 |
| Read | - | - | - | - | ON | - |
| Ar flow rate/ml min−1 | 300 | 300 | 300 | 300 | 0 | 300 |
| Analyte | Atomization | Chemical modifier | TPyr/°C | TAtom/°C |
|---|---|---|---|---|
| Cadmium | Wall | None | 250 | 2000 |
| Platform | None | 500 | 1800 | |
| Platform | NH4H2PO4 + Mg(NO3)2 | 1200 | 2000 | |
| Platform | Oxalic acid | 500 | 1600 | |
| Lead | Wall | None | 400 | 1800 |
| Platform | None | 1000 | 2200 | |
| Platform | NH4H2PO4 + Mg(NO3)2 | 1400 | 2400 | |
| Platform | Pd(NO3)2 + Ascorbic acid | 1300 | 2400 | |
| Copper | Wall | None | 800 | 2300 |
| Platform | None | 1500 | 2800 | |
| Platform | Pd(NO3)2 + Mg(NO3)2 | 1500 | 2800 | |
| Platform | Oxalic acid | 1500 | 2800 | |
| Iron | Wall | None | 800 | 2300 |
| Platform | None | 1600 | 2800 | |
| Manganese | Wall | None | 800 | 2300 |
| Platform | None | 1400 | 2600 | |
| Platform | Pd(NO3)2 + Mg(NO3)2 | 1600 | 2600 | |
| Platform | Oxalic acid | 1500 | 2600 |
| Element | Wavelength/nm | Slit width/nm | Lamp current/mA | Time constant/s | Integration time/s |
|---|---|---|---|---|---|
| Cd | 228.8 | 0.5 | 6 | 0.05 | 3.0 |
| Pb | 283.3 | 1.0 | 4 | 0.05 | 3.0 |
| Cu | 327.4 | 0.5 | 4 | 0.05 | 3.0 |
| Fe | 248.3 | 0.2 | 10 | 0.05 | 3.0 |
| Mn | 279.5 | 0.2 | 8 | 0.05 | 3.0 |
ICP-AES analyses were performed using a Varian (Springvale, Australia) Liberty 100 atomic emission spectrometer, while ICP-MS measurements were carried out on a Perkin-Elmer (Shelton, CT, USA) SCIEX Elan 6000 ICP mass spectrometer, both working under the recommended operating conditions.
Reagents
1 mg ml−1 and 10 mg ml−1 standard solutions of Na, K, Mg and Ca in 0.5 mol l−1 HNO3 were obtained from BDH Chemicals (Poole, Dorset, UK) and used as matrix stock solutions. 1 mg ml−1 standard solutions of Cd, Pb, Cu, Fe and Mn, in 0.5 mol l−1 HNO3, were also obtained from BDH Chemicals and used as the analyte stock solutions. Synthetic samples were prepared daily by the addition of the appropriate aliquots of the above-mentioned standard solutions and by dilution with Milli-Q water (Millipore, El Paso, TX, USA). All solutions were made up in 0.1 mol l−1 HNO3 of suprapure grade quality (Merck, Darmstadt, Germany).Chemical modifier solutions were prepared as follows. 0.2 mol l−1 oxalic acid was obtained by dissolving 2.5214 g of (COOH)2·2H2O (Carlo Erba, Milan, Italy) in 100 ml of Milli-Q water. The ammonium dihydrogenphosphate–magnesium nitrate modifier was prepared by dissolving 1.2112 g of NH4H2PO4 of suprapure grade (Merck, Darmstadt, Germany) and 0.0864 g of Mg(NO3)2·6H2O of suprapure grade (Merck, Darmstadt, Germany) in 25 ml of Milli-Q water. The palladium nitrate–magnesium nitrate modifier was prepared by mixing equal volumes of a solution containing 3000 mg l−1 of Pd and 2000 mg l−1 of Mg(NO3)2. Chloride-free palladium matrix modifier solution, 99.999% containing 2% Pd in 1 wt.% HNO3, was purchased from Aldrich (Milwaukee, WI, USA). 0.2 mol l−1 ascorbic acid was prepared by dissolving 3.5226 g of the salt (Carlo Erba, Milan, Italy) in 100 ml of Milli-Q water.
1 mol l−1 ammonium acetate buffer was prepared by combining 7.7 ml of 25% NH4OH of suprapure grade (Merck, Darmstadt, Germany) with 5.7 ml of glacial acetic acid of suprapure grade (Merck, Darmstadt, Germany) and diluting to 100 ml. The acidity was adjusted to pH 5.5 by dropwise addition of acetic acid or NH4OH.
The sea-water reference materials NASS-5 and CASS-3 were purchased from the National Research Council Canada. The pH of each sample was adjusted to 6–7 by addition of 3.6 g l−1 sodium carbonate of suprapure grade (Merck, Darmstadt, Germany).
Analytical procedure
A scheme of the analytical procedure is presented in Fig. 1. The preconcentration of heavy metals from sea-water was obtained by delivering each sample through a micro-column filled with a iminodiacetic resin (size range: 150–300 µm; exchange capacity: 2.9 meq g−1). Ready-to-use micro-columns (IC-Chelate) were purchased from Alltech (Deerfield, IL, USA). Up to 24 samples were simultaneously prepared by using a multi-channel peristaltic pump (Technicon, Dublin, Ireland). | ||
| Fig. 1 Schematic of analytical procedure. | ||
The resin and the connecting tubes were cleaned by a flow of 1 M nitric acid (500 ml, 0.42 ml min−1). The concentration of the considered elements after this step was checked by ICP-MS and ETAAS measurements and values above the instrumental detection limits were found for all the elements. Then, the resin was treated with 1 M ammonium acetate buffer (pH 5.5, 0.42 ml min−1, 20 min), in order to replace the H+ ions with NH4+ ions. Contamination due to this step was again found to be negligible.
Sea-water samples and blanks were delivered through the columns (100 ml, 0.42 ml min−1). After, the samples were eluted with 1 M nitric acid, 1–1.5 ml of solution were collected into pre-cleaned sampling cups, and analysed by ETAAS (for trace element determination) and ICP-AES (for major element determination).
In some experiments, pre-elution with 1 M ammonium acetate (pH 5.5, 0.42 ml min−1, 30 min) was performed in order to remove the major elements before the ETAAS determination.
Multivariate experiments and data processing
The multivariate study of matrix effects was performed according to a method previously reported.24 The adopted experimental design is presented in Table 4. According to this, the experiments are performed on a spherical domain, at the vertices of the hypercube corresponding to a 25−1 fractional design (points 1–16), at the so-called “star points” (points 17–26) and at the centre of the domain (points 27–32). The replicates at the centre point give the estimate of the experimental variance, taking into account both the instrumental precision and the sample preparation procedure.| Run | Na | K | Ca | Mg | Me |
|---|---|---|---|---|---|
| 1 | −1 | −1 | −1 | −1 | +1 |
| 2 | +1 | −1 | −1 | −1 | −1 |
| 3 | −1 | +1 | −1 | −1 | −1 |
| 4 | +1 | +1 | −1 | −1 | +1 |
| 5 | −1 | −1 | +1 | −1 | −1 |
| 6 | +1 | −1 | +1 | −1 | +1 |
| 7 | −1 | +1 | +1 | −1 | +1 |
| 8 | +1 | +1 | +1 | −1 | −1 |
| 9 | −1 | −1 | −1 | +1 | −1 |
| 10 | +1 | −1 | −1 | +1 | +1 |
| 11 | −1 | +1 | −1 | +1 | +1 |
| 12 | +1 | +1 | −1 | +1 | −1 |
| 13 | −1 | −1 | +1 | +1 | +1 |
| 14 | +1 | −1 | +1 | +1 | −1 |
| 15 | −1 | +1 | +1 | +1 | −1 |
| 16 | +1 | +1 | +1 | +1 | +1 |
| 17 | −√5 | 0 | 0 | 0 | 0 |
| 18 | +√5 | 0 | 0 | 0 | 0 |
| 19 | 0 | −√5 | 0 | 0 | 0 |
| 20 | 0 | +√5 | 0 | 0 | 0 |
| 21 | 0 | 0 | −√5 | 0 | 0 |
| 22 | 0 | 0 | +√5 | 0 | 0 |
| 23 | 0 | 0 | 0 | −√5 | 0 |
| 24 | 0 | 0 | 0 | +√5 | 0 |
| 25 | 0 | 0 | 0 | 0 | −√5 |
| 26 | 0 | 0 | 0 | 0 | +√5 |
| 27–32 | 0 | 0 | 0 | 0 | 0 |
By fixing the ranges of the variables and the scale (a logarithmic scale for the interfering elements and a linear scale for the analytes), the coded values were replaced by real values and the experimental plan was obtained (levels of the variables are shown in Table 5). According to this, each solution was prepared and analysed by ETAAS, using a calibration curve based on standard solutions in 0.1 mol l−1 HNO3. Pyro-coated tubes with L'Vov platforms were used.
| Variable | Level | ||||
|---|---|---|---|---|---|
| −√5 | −1 | 0 | +1 | +√5 | |
| Na | 0.5 | 4.1 | 22.4 | 122.5 | 1000 |
| K | 0.5 | 4.1 | 22.4 | 122.5 | 1000 |
| Mg | 0.5 | 4.1 | 22.4 | 122.5 | 1000 |
| Ca | 0.5 | 4.1 | 22.4 | 122.5 | 1000 |
| Cd | 0 | 0.55 | 1 | 1.45 | 2 |
| Cu | 0 | 2.8 | 5 | 7.2 | 10 |
| Fe | 0 | 5.5 | 10 | 14.5 | 20 |
| Mn | 0 | 4.2 | 7.5 | 10.8 | 15 |
| Pb | 0 | 5.5 | 10 | 14.5 | 20 |
Data were processed by performing a multi-linear regression (MLR) analysis in which the integrated absorbance was considered as an independent variable and the analytical concentrations of both trace and major elements as the dependent variables.
The quality of the MLR analyses was tested by performing the “cross-validation” procedure.25 Each experiment was removed from the training set and the model was re-calculated. Then, the predicted value of the missing experiment was computed by the new model and compared with the true one. This procedure was repeated for all the experiments and the explained variance (EV) was calculated:{*BLOB:S*}
 | (1) |
Multiple linear regression analyses and the other statistical calculations were performed using the package of programs, Parvus 1.2.26
Results and discussion
Due to the interaction between the imminodiacetic groups' ion pairs and the electron free d-orbitals of the transition element ions, the resin is able to strongly bind the heavy metals, and their complete recovery from complex matrices is hence obtained. However, the sorption mechanism is not specific and other elements, mainly bivalent ions, can be similarly retained, especially if they are present at high concentration levels.In the case of trace element preconcentration from sea-water, major elements such as sodium, potassium, calcium and magnesium are partially retained by the resin and are co-eluted with the analytes (Fig. 2). Therefore, matrix interferences are expected in the following ETAAS determination.
 | ||
| Fig. 2 Matrix composition before and after preconcentration step. | ||
Multivariate quantification of matrix effects
In order to obtain a multivariate evaluation of the interfering effects due to sodium, potassium, calcium and magnesium on the electrothermal atomization of cadmium, lead, copper, iron and manganese, the MLR method was applied. The procedure has been extensively described previously24 and has been successfully applied to quantify the interferences in atomic absorption,24,27–29 emission30 and plasma mass31 spectrometry.According to the method, the following relationship between the analytical signal and the concentrations of both analyte and matrix elements is considered:
 | (2) |
| Coefficient | Factor | QA (Cd) | QA (Pb) | QA (Cu) | QA (Fe) | QA (Mn) |
|---|---|---|---|---|---|---|
| Intercept | ||||||
| b0 | mean | 0.074 ± 0.005 | 0.044 ± 0.001 | 0.035 ± 0.003 | 0.132 ± 0.007 | 0.344 ± 0.014 |
| Main effects | ||||||
| b1 | (Na) | −0.004 ± 0.003 | 0.017 ± 0.012 | |||
| b2 | (K) | 0.006 ± 0.004 | −0.002 ± 0.001 | 0.006 ± 0.003 | 0.009 ± 0.006 | |
| b3 | (Mg) | −0.005 ± 0.004 | 0.002 ± 0.001 | −0.005 ± 0.003 | 0.026 ± 0.006 | 0.023 ± 0.015 |
| b4 | (Ca) | −0.007 ± 0.004 | −0.002 ± 0.001 | 0.004 ± 0.003 | 0.007 ± 0.006 | |
| b5 | (Me) | 0.019 ± 0.004 | 0.018 ± 0.001 | 0.013 ± 0.003 | 0.054 ± 0.006 | 0.130 ± 0.012 |
| Quadratic effects | ||||||
| b11 | (Na)2 | |||||
| b22 | (K)2 | |||||
| b33 | (Mg)2 | −0.002 ± 0.001 | 0.011 ± 0.005 | 0.053 ± 0.014 | ||
| b44 | (Ca)2 | |||||
| b44 | (Me)2 | |||||
| Two-factors interactions | ||||||
| b12 | (Na)(K) | −0.008 ± 0.005 | 0.002 ± 0.001 | −0.005 ± 0.004 | −0.023 ± 0.008 | |
| b13 | (Na)(Mg) | −0.009 ± 0.004 | −0.019 ± 0.008 | |||
| b14 | (Na)(Ca) | 0.005 ± 0.004 | 0.013 ± 0.008 | |||
| b15 | (Na)(Me) | 0.005 ± 0.004 | 0.016 ± 0.015 | |||
| b23 | (K)(Mg) | 0.014 ± 0.008 | ||||
| b24 | (K)(Ca) | −0.009 ± 0.004 | −0.020 ± 0.008 | |||
| b25 | (K)(Me) | 0.018 ± 0.008 | ||||
| b34 | (Mg)(Ca) | 0.006 ± 0.005 | −0.010 ± 0.004 | −0.018 ± 0.008 | ||
| b35 | (Mg)(Me) | 0.009 ± 0.008 | ||||
| b45 | (Ca)(Me) | −0.016 ± 0.015 | ||||
| Multiple linear regression coefficient | ||||||
| 0.93 | 0.98 | 0.95 | 0.99 | 0.98 | ||
| Cross validation Explained Variance | ||||||
| 71% | 94% | 69% | 85% | 94% | ||
Table 6 contains a great deal of information and shows how complex the relationships are between interference effects and matrix composition. In general, all the concomitant elements showed significant effects, which can be positive or negative depending on the analyte. The strongest effect is that due to magnesium, mainly on the copper, iron and manganese determination. Since significant quadratic terms were deduced, this effect increases sharply with increasing concentration. Finally, significant two-term interactions were deduced and must be considered in order to obtain a complete quantification of matrix effects.
By inserting the concentration values of major elements after the preconcentration step (Fig. 2) into the models, significant matrix-induced signal variations are predicted, even if a 1∶10 dilution of the matrix is considered (Table 7).
| Analyte | Signal variation (%) | |
|---|---|---|
| Matrix 1∶10 | Matrix 1∶1 | |
| Cd | −69% | −53% |
| Pb | −58% | −62% |
| Cu | −36% | −92% |
| Fe | −24% | 26% |
| Mn | 4% | 69% |
Reduction of matrix effects by chemical modification
Chemical modification is considered to be the more general and effective approach to reduce matrix effects in ETAAS. Several modifiers have been proposed for cadmium, lead, copper and manganese determination in the presence of alkaline and alkaline earth elements, mainly as chlorides.32–40 However, iron determination is usually performed without any chemical modifier, as suggested by Sturgeon et al.41 The effectiveness of the chemical modifiers in decreasing the interference effects that have been previously quantified was checked on two test solutions. The first solution simulated a real sample after the preconcentration step, while the second contained the same matrix elements, but at concentrations 10-times lower. In each case, the signal variation caused by the considered matrices was evaluated as a function of the pyrolysis temperature. Graphite tubes with L'Vov platform were used exclusively. The results obtained with the undiluted matrix are reported in Figs. 3–7. | ||
| Fig. 3 Cadmium AAS signal variation due to the major elements still present after the preconcentration step. Symbols: ●, no modifier; ■, NH4H2PO4 + Mg(NO3)2; ◆, oxalic acid. | ||
 | ||
| Fig. 4 Lead AAS signal variation due to the major elements still present after the preconcentration step. Symbols: ●, no modifier; ■, NH4H2PO4 + Mg(NO3)2; ◆, Pd(NO3)2 + ascorbic acid. | ||
 | ||
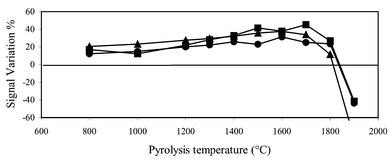
| Fig. 5 Copper AAS signal variation due to the major elements still present after the preconcentration step. Symbols: ●, no modifier; ■, Pd(NO3)2 + Mg(NO3)2; ◆, oxalic acid. | ||
 | ||
| Fig. 6 Manganese AAS signal variation due to the major elements still present after the preconcentration step. Symbols: ●, no modifier; ■, Pd(NO3)2 + Mg(NO3)2; ▲, oxalic acid. | ||
 | ||
| Fig. 7 Iron AAS signal variation due to the major elements still present after the preconcentration step. | ||
In the absence of a chemical modifier, a substantial signal decrease was noted for cadmium (Fig. 3), lead (Fig. 4) and copper (Fig. 5), which is in good agreement with the matrix effects predicted by the models (Table 7). Also the interfering effects due to the 1∶10 diluted matrix were significant. A further signal decrease was observed at pyrolysis temperatures higher than 600°C for cadmium and 1000°C for lead and copper, due to the analyte loss from the graphite atomizer.
In order to reduce the observed interferences, several modifiers were considered, according to literature data.32–40 For cadmium determination, the use of oxalic acid was not efficient especially in the case of the undiluted matrix (Fig. 3). An excellent reduction of matrix effects was obtained with the NH4H2PO4 + Mg(NO3)2 mixture. The maximum pyrolysis temperature without loss of analyte increased from 600 to 1300°C and signal variation decreased from 40% to 5% and from 60% to 10% for the diluted and undiluted matrix, respectively.
A significant reduction of matrix effects was obtained for lead determination, both with the use of Pd(NO3)2 + ascorbic acid and of NH4H2PO4 + Mg(NO3)2
(Fig. 4). In the presence of these modifiers, the maximum pyrolysis temperature increased from 1000 to 1300°C and 1400°C, respectively, and the signal variation was less than 10% for both the matrices. Chemical modification was efficient also for copper determination (Fig. 5). Oxalic acid and the palladium nitrate–magnesium nitrate mixture allowed the use of higher pyrolysis temperatures (1500°C) and a significant decrease in matrix effects was hence obtained. The same modifiers were tested also for manganese determination, but strong signal variations
caused by the matrix were observed (Fig. 6). Ten-times dilution of the matrix led to a decrease of matrix effects, also without a chemical modifier. Again, these results are in good agreement with the prediction based on the multivariate study (Table 7). Finally, a signal increase of 40% due to the undiluted matrix was found for iron in the 800–1800°C pyrolysis temperature range (Fig. 7). In the presence of the diluted matrix, the signal variation was less than 10%.
A comparison of analytical performance for all the determinations with and without chemical modifiers is summarized in Table 8 (results obtained for tube atomization are also reported). Taking into account thermal stabilization, sensitivity (characteristic mass) and interference tolerance, the optimal conditions were chosen (in bold).
| Analyte | Atomization | Chemical modifier | TPyr max/°C | m0/pg | Signal variation (%) | |
|---|---|---|---|---|---|---|
| Matrix 1∶10 | Matrix 1∶1 | |||||
| Cadmium | Wall | None | 250 | 0.85 | 0 | −32 |
| Platform | None | 500 | 0.62 | −40 | −58 | |
| Platform | NH4H2PO4 + Mg(NO3)2 | 1200 | 0.63 | 5 | −10 | |
| Platform | Oxalic acid | 500 | 0.50 | −22 | −71 | |
| Lead | Wall | None | 400 | 13.5 | −17 | −29 |
| Platform | None | 1000 | 11.1 | −39 | −52 | |
| Platform | NH4H2PO4 + Mg(NO3)2 | 1400 | 13.5 | −7 | −6 | |
| Platform | Pd(NO3)2 + Ascorbic acid | 1300 | 15.2 | −2 | −4 | |
| Copper | Wall | None | 800 | 11.9 | −53 | −69 |
| Platform | None | 1500 | 18.0 | −31 | −37 | |
| Platform | Pd(NO3)2 + Mg(NO3)2 | 1500 | 25.1 | −8 | −1 | |
| Platform | Oxalic acid | 1500 | 19.6 | −8 | 0 | |
| Iron | Wall | None | 800 | 6.7 | 4 | 55 |
| Platform | None | 1600 | 6.7 | 7 | 52 | |
| Manganese | Wall | None | 800 | 2.9 | 5 | 59 |
| Platform | None | 1400 | 2.4 | 8 | 26 | |
| Platform | Pd(NO3)2 + Mg(NO3)2 | 1600 | 3.7 | 2 | 38 | |
| Platform | Oxalic acid | 1500 | 3.0 | 6 | 36 | |
Under these conditions, the analysis of the certified reference materials NASS-5 was performed (Table 9). A satisfactory accuracy was observed for cadmium and lead, while manganese and iron concentrations were slightly over-estimated, due to the matrix effects previously outlined. The poor copper recovery is probably related to the unfavourable competition of iminodiacetic active groups with organically bound copper.
| Analyte | Found/µg l−1 | Certified/µg l−1 |
|---|---|---|
| Cd | 0.019 ± 0.003 | 0.023 ± 0.003 |
| Pb | 0.006 ± 0.001 | 0.008 ± 0.005 |
| Cu | 0.204 ± 0.004 | 0.297 ± 0.046 |
| Fe | 0.263 ± 0.032 | 0.207 ± 0.035 |
| Mn | 1.149 ± 0.017 | 0.919 ± 0.057 |
Reduction of matrix effects by matrix separation
Alkali and alkaline earth elements can be efficiently separated from the analytes by elution with ammonium acetate.42 The selective elution was carried out with 1 M ammonium acetate at pH 5.5, at a flow rate of 0.42 ml min−1 for 30 min.Concentrations of alkali and alkali earth elements after the pre-elution step were checked by ICP-AES, and it was found that the matrix element removal was complete. The accuracy of the procedure was finally confirmed by analysis of the certified reference materials CASS-3 and NASS-5 (Table 10). As stated above, the under-estimation of the copper concentration is probably due to the preconcentration procedure and not to matrix effects in the ETAAS determination. For all the other analytes, a good accuracy was noted, proving the efficient elimination of the matrix effects.
| Analyte | NASS-5 | CASS-3 | ||
|---|---|---|---|---|
| Found/µg l−1 | Certified/µg l−1 | Found/µg l−1 | Certified/µg l−1 | |
| a Spiked sea-water. | ||||
| Cd | 0.025 ± 0.001 | 0.023 ± 0.003 | 0.031 ± 0.001 | 0.030 ± 0.005 |
| Pba | 0.124 ± 0.003 | 0.107 ± 0.005 | 0.078 ± 0.004 | 0.099 ± 0.005 |
| Cu | 0.119 ± 0.004 | 0.297 ± 0.046 | 0.224 ± 0.013 | 0.517 ± 0.062 |
| Fe | 0.235 ± 0.015 | 0.207 ± 0.035 | 1.36 ± 0.16 | 1.26 ± 0.17 |
| Mn | 0.960 ± 0.012 | 0.919 ± 0.057 | 2.64 ± 0.17 | 2.51 ± 0.36 |
Acknowledgements
This work was financially supported by MURST-COFIN 2000, Fondi di Ateneo, Università di Genova.References
- Z. Fang, M. Sperling and B. Welz, J. Anal. At. Spectrom., 1990, 5, 639 RSC.
- M. Sperling, X. Yin and B. Welz, J. Anal. At. Spectrom., 1991, 6, 295 RSC.
- V. Porta, O. Abollino, E. Mentasti and C. Sarzanini, J. Anal. At. Spectrom., 1991, 6, 119 RSC.
- Z. Liu and S. Huang, Spectrochim. Acta, Part B, 1995, 50, 197 CrossRef.
- D. Colbert, K. S. Johnson and K. H. Coale, Anal. Chim. Acta, 1998, 377, 255 CrossRef CAS.
- X. Yan and F. Adams, J. Anal. At. Spectrom., 1997, 12, 459 RSC.
- M. Sperling, X. Yan and B. Welz, Spectrochim. Acta, Part B, 1996, 51, 1891 CrossRef.
- K. Benkhedda, H. G. Infante, E. Ivanova and F. Adams, J. Anal. At. Spectrom., 2000, 15, 429 RSC.
- F. M. Fernandez, J. D. Stripeikis, M. B. Tudino and O. E. Troccoli, Analyst, 1997, 122, 679 RSC.
- M. M. Silva, F. J. Krug, P. V. Oliveira, J. A. Nobrega, B. F. Reis and D. A. G. Penteado, Spectrochim. Acta, Part B, 1996, 51, 1925 CrossRef.
- F. M. Fernandez, J. D. Stripeikis, M. B. Tudino and O. E. Troccoli, Analyst, 1997, 122, 679 RSC.
- F. Baffi, A. M. Cardinale and R. Bruzzone, Anal. Chim. Acta, 1992, 270, 79 CrossRef CAS.
- S.-C. Pai, T. H. Fang, C.-T. A. Chen and K.-L. Jeng, Mar. Chem., 1990, 29, 295 CrossRef CAS.
- F. M. Fernandez, M. B. Tudino and O. E. Troccoli, J. Anal. At. Spectrom., 2000, 15, 687 RSC.
- Y.-H. Sung, Z.-S. Liu and S.-D. Huang, J. Anal. At. Spectrom., 1997, 12, 841 RSC.
- Y.-H. Sung, Z.-S. Liu and S.-D. Huang, Spectrochim. Acta, Part B, 1997, 52, 766 CrossRef.
- M. T. S. Cordero, E. I. V. Alonso, P. C. Rudner, A. G. de Torres and J. M. C. Pavon, J. Anal. At. Spectrom., 1999, 14, 1033 RSC.
- S. Sella, R. E. Sturgeon, S. N. Willie and R. C. Campos, J. Anal. At. Spectrom., 1997, 12, 1281 RSC.
- K. Akatsuka and I. Atsuya, Fresenius' J. Anal. Chem., 1987, 329, 453 CAS.
- S. Nakashima, R. E. Sturgeon, S. N. Willie and S. S. Berman, Anal. Chim. Acta, 1988, 207, 291 CrossRef CAS.
- H. Niskavaara and E. Kontas, Anal. Chim. Acta, 1990, 231, 273 CrossRef CAS.
- R. K. Skogerboe, W. A. Hanagan and H. E. Taylor, Anal.Chem., 1985, 57, 2815 CrossRef CAS.
- J. Komarek and J. Holy, Spectrochim. Acta, Part B, 1999, 54, 733 CrossRef.
- M. Grotti, R. Leardi and R. Frache, Anal. Chim. Acta, 1998, 376, 293 CrossRef CAS.
- M. Stone, J. R. Stat. Soc., Ser. B, 1974, 36, 111 Search PubMed.
- M. Forina, S. Lanteri, C. Armanino, R. Leardi and G. Drava, PARVUS 1.2 An extendable package of programs for data explorative analysis, classification and regression analysis, 1995 Search PubMed.
- M. Grotti, E. Magi and R. Leardi, Anal. Chim. Acta, 1996, 327, 47 CrossRef CAS.
- M. Grotti, R. Leardi, C. Gnecco and R. Frache, Spectrochim. Acta, Part B, 1999, 54, 845 CrossRef.
- M. Grotti, M. L. Abelmoschi, F. Soggia, F. Tiberiade and R. Frache, Spectrochim. Acta, Part B, 2000, 55, 1847 CrossRef.
- M. Grotti, E. Magi and R. Frache, J. Anal. At. Spectrom., 2000, 15, 89 RSC.
- M. Grotti, C. Gnecco and F. Bonfiglioli, J. Anal. At. Spectrom., 1999, 14, 1171 RSC.
- B. Welz, G. Schlemmer and J. R. Mudakavi, J. Anal. At. Spectrom., 1988, 3, 695 RSC.
- J. Y. Cabon and A. Le Bihan, J. Anal. At. Spectrom., 1992, 7, 383 RSC.
- C.-R. Lan, Analyst, 1993, 118, 189 RSC.
- P.-G. Su and S.-D. Huang, Spectrochim. Acta, Part B, 1998, 53, 699 CrossRef.
- H. Chuang and S.-D. Huang, Spectrochim. Acta, Part B, 1994, 49, 283 CrossRef.
- J. Y. Cabon and A. Le Bihan, Spectrochim. Acta, Part B, 1995, 50, 1703 CrossRef.
- P. Bermejo-Barrera, J. Moreda-Piñeiro, A. Moreda-Piñeiro and A. Bermejo-Barrera, J. Anal. At Spectrom., 1998, 13, 777 RSC.
- O. Acar, A. R. Turker and Z. Kilic, Talanta, 1999, 49, 135 CrossRef CAS.
- M.-S. Chang and S.-D. Huang, Talanta, 2000, 51, 373 CrossRef CAS.
- R. E. Sturgeon, S. S. Berman, A. Desaulniers and D. S. Russell, Anal. Chem., 1979, 51, 2364 CrossRef CAS.
- H. M. Kingston, I. L. Barnes, T. J. Brady and T. C. Rains, Anal. Chem., 1978, 50, 2064 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |