Control of spectral and non-spectral interferences in the determination of thallium in river and marine sediments using solid sampling electrothermal atomic absorption spectrometry
Maria Goreti R. Valea, Marcia M. Silvaa, Bernhard Welz*b and René Nowkac
aInstituto de Química, Universidade Federal do Rio Grande do Sul, Avenida Bento Gonçalves 9500, 91501-970, Porto Alegre–RS, Brazil
bDepartamento de Química, Universidade Federal de Santa Catarina, 88040-900, Florianópolis–SC, Brazil. E-mail: wbernard@matrix.com.br
cAnalytik Jena AG, Konrad-Zuse-Str. 1, D-07745, Jena, Germany
First published on 29th November 2001
Abstract
The direct analysis of solid samples using electrothermal (graphite furnace) atomic absorption spectrometry (ETAAS) has been investigated for the determination of thallium in river and marine sediment reference materials, because complete digestion of sediment samples requires the use of hydrofluoric acid and/or an alkaline fusion, and the extraction with aqua regia might be incomplete and cause interferences in the determination of thallium. The determination of thallium in river sediments using direct solid sampling ETAAS was straight forward, and could be carried out with good accuracy even without the use of a chemical modifier or calibration against aqueous standards. The analysis of marine sediments, in contrast, proved to be extremely difficult due to severe spectral and non-spectral interferences. The latter ones were caused by the relatively high chloride content of marine sediments, compared to river sediments, and could eventually be controlled by the addition of ammonium nitrate as a chemical modifier together with ruthenium as a permanent modifier. The spectral interference could only be overcome with Zeeman-effect background correction, and was most likely caused by sulfate. After optimization of the procedure, thallium in marine sediment reference materials could be determined by calibration against a certified river sediment reference material. According to the experience gained with river sediments, it might be assumed that aqueous standards could equally be used for calibration; this approach, however, was not further investigated. The results were in good agreement with non-certified ‘information’ values for thallium and with results obtained by ICP-MS using electrothermal vaporization and isotope dilution calibration. A characteristic mass of 13 pg was obtained, and the limit of detection of the proposed method, based on the zero-mass response (three times the standard deviation of 10 atomization cycles with empty platforms), was around 0.02 µg g−1 Tl.
Introduction
Thallium and many of its compounds exhibit a relatively high toxicity. Even a few milligrams can cause severe chronic intoxication, resulting, for example, in loss of hair, nervous disease, impaired vision and growth retardation.1 Thallium is also toxic for many animals, plants and microorganisms, and may be enriched in the food chain.2 Thallium is usually found only in small amounts in animals and plants, but larger quantities may be contained in stack gases and airborne dust, particularly from cement works,1 which will end up in soils and sediments through atmospheric deposition. It would, therefore, be useful to determine this element routinely when soils and sediments are analysed. Unfortunately, however, only very few reference materials have a certified value for thallium, making validation of the methods difficult. It is not clear if this is due to a lack of interest in this element, or to analytical difficulties in its determination.It is well known that electrothermal (graphite furnace) atomic absorption spectrometry (ETAAS), one of the most suitable techniques for this type of analysis, suffers from a persistent chloride interference,3 which cannot be completely eliminated even when the stabilized temperature platform furnace (STPF) concept4 is used. L'vov5 stated that formation of gaseous monohalides is the most frequent source of non-spectral interferences in ETAAS, and the influence of chloride on thallium is a typical example of this interference. For eliminating or diminishing this effect, L'vov proposed to increase the atomization temperature by vaporizing the sample from a graphite platform, and to bind the free chlorine into molecules by the addition of lithium. Jackson and co-workers6,7 found that this interference, i.e., the formation of thallium chloride, occurred between condensed-phase species during the pyrolysis stage. Bulska and Ortner8 showed that chloride forms stable graphite intercalation compounds, which might, at least in part, be responsible for this interference.
Slavin and co-workers have shown in several publications that atomization from a L'vov platform substantially reduced the interference from sodium chloride on thallium.9 The addition of sulfuric acid further increased the range of interference-free determination.10,11 Welz et al.12 confirmed the finding of L'vov5 that the addition of lithium reduced the chloride interference on thallium but could not remove it completely. Shan et al.13 developed a method for the determination of thallium in waste water by using palladium as a modifier which allowed the pyrolysis temperature to be raised to 1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C. Welz et al.14 found that a mixture of palladium and magnesium nitrates resulted in a higher ruggedness and better tolerance for chloride in the determination of
thallium in airborne particulate matter. They also found it necessary to use Zeeman-effect background correction in order to deal with the high non-specific absorption. Voth-Beach and Shrader15 also found that the performance of palladium alone as a modifier was strongly affected by the sample matrix and that the addition of a reducing agent provided for more consistent performance. Their results were seriously impaired, however, because these authors used atomization from the tube wall and not from a platform, which made the analysis more susceptible to interferences and less comparable to the results of others.
°C. Welz et al.14 found that a mixture of palladium and magnesium nitrates resulted in a higher ruggedness and better tolerance for chloride in the determination of
thallium in airborne particulate matter. They also found it necessary to use Zeeman-effect background correction in order to deal with the high non-specific absorption. Voth-Beach and Shrader15 also found that the performance of palladium alone as a modifier was strongly affected by the sample matrix and that the addition of a reducing agent provided for more consistent performance. Their results were seriously impaired, however, because these authors used atomization from the tube wall and not from a platform, which made the analysis more susceptible to interferences and less comparable to the results of others.
Welz et al.16 confirmed that the stabilizing power of the palladium modifier could be improved significantly for the determination of thallium in the presence of high chloride concentrations when it was reduced before sample injection. For the determination of thallium in seawater and urine, it was even found necessary to use hydrogen as a reducing purge gas in addition to the pre-reduction of the palladium modifier. Yang and Smeyers-Verbeke17 preferred to use a mixture of palladium and ammonium nitrates for the determination of thallium in serum and urine; a modifier that had been proposed by Yin et al.18 to overcome chloride interferences in the determination of cadmium. Manning and Slavin19 confirmed that palladium is an effective modifier for the determination of thallium. In the presence of high chloride concentrations, however, they preferred
to abandon the pyrolysis stage and the modifier completely, and to use Zeeman-effect background correction in order to cope with the high non-specific absorption that was observed under these conditions. López-García et al.20 followed this example, working without a modifier, and used a ‘modified drying stage’ at 400°C for the determination of thallium in soils and sediments using slurry sampling ETAAS.
Not only the ETAAS determination of thallium is impaired by this chloride interference, but also inductively coupled plasma mass spectrometry (ICP-MS) with electrothermal vaporization (ETV) is strongly influenced. Even isotopic dilution calibration cannot compensate for the volatilization of the analyte as TlCl in the pyrolysis stage.21 In addition to chloride, sulfate appears to interfere as well with the determination of thallium in ETAAS, and Matsusaki and Oishi22 used a mixture of the ammonium salt of the ethylene diamine acetic acid (EDTA) and nickel nitrate to overcome this interference. Zendelovska and Stafilov,23 in addition to the chloride interference, found that iron tended to increase the absorbance of thallium in sulfide minerals.
The total analyte concentration in a sediment or soil can only be determined after fusion with sodium carbonate24 or lithium metaborate,25–27 or an acid digestion in the presence of hydrofluoric and frequently also perchloric acid.28–30 These procedures are time-consuming and associated with the risks of analyte loss and/or contamination. For these reasons, an extraction with boiling aqua regia under reflux is frequently preferred over a total digestion.31–33 Such an extraction, however, may be incomplete, as has been shown by Waidmann et al.,34 who reported that only 43–86% of the total amount of thallium present in sediment samples was found after extraction with aqua regia. Although it has been demonstrated that such extracts can be handled by ETAAS, particularly in the presence of permanent chemical modifiers,35 it was suspected that, in the case of thallium, the aqua regia medium would aggravate the chloride interference problem. This was confirmed by Zendelovska and Stafilov,23 who found that it is not possible to determine thallium directly from solutions obtained by dissolution of minerals in mineral acids (HNO3 + HCl).
The goal of the present work was to investigate the possibility of using direct solid sampling (SS) ETAAS for the determination of thallium in river and marine sediments on a routine basis, in order to avoid the above-described problems. The somewhat lower precision of solid sampling, compared with the analysis of solutions, was considered insignificant in this case, particularly in view of the natural inhomogeneity of environmental samples such as sediments, and the associated difficulty in obtaining a truly representative sample from an environmental situation. Moreover, this loss in precision might be more than compensated by a gain in accuracy of the results, as, besides some additional grinding, essentially no sample preparation is required and no new matrix components are introduced. Part of the investigation was devoted to the problem of calibration, which can, on occasion, be more difficult in solid sample analysis compared with solution analysis. The use of appropriate modifiers – if required – was another topic of this investigation, and the use of a permanent modifier was a special goal, as this was found to significantly simplify the direct analysis of solid samples.36 A comparison of continuum-source and Zeeman-effect background correction for the determination of thallium in sediments was also part of the investigation.
Experimental
Instrumentation
Most of the measurements were carried out using an AAS 5 EA atomic absorption spectrometer (Analytik Jena AG, Jena, Germany), equipped with a transversely heated graphite tube atomizer, an SSA 5 manual solid sampling accessory and continuum-source background correction. Some of the experiments were carried out using a ZEEnit 60 atomic absorption spectrometer (Analytik Jena AG, Jena, Germany), equipped with a transversely heated graphite tube atomizer with Zeeman-effect background correction in the transverse configuration and with an SSA 6Z manual solid sampling accessory. A NARVA hollow cathode lamp for thallium (G.L.E., Berlin, Germany) was used as the radiation source. The lamp current was set to 6.0 mA, and the primary resonance line at 276.8 nm was used for all determinations with a spectral bandwidth of 0.5 nm. The ZEEnit 60 was operated in the two-field mode with a maximum field strength of 1.0 Tesla. Integrated absorbance (peak area) was used exclusively for quantification.The experiments with the AAS 5 EA instrument were carried out using pyrolytic graphite-coated solid sampling graphite tubes without a dosing hole (Analytik Jena AG, Part No. 07-8130325), whereas conventional pyrolytic graphite-coated tubes with a dosing hole, but without a platform, were used for the experiments with the ZEEnit 60 instrument. the samples were introduced on solid sampling platforms (Analytik Jena AG, Part No. 407-A81.312) using a pre-adjusted pair of tweezers, which is part of the SSA 5 and SSA 6Z manual solid sampling accessories. Solid samples were weighed directly onto the platforms using an M2P microbalance (Sartorius, Göttingen, Germany). The accurate sample mass, typically around 1 mg, was automatically transmitted to the instrument's computer to calculate the ‘normalized integrated absorbance’ (integrated absorbance calculated for 1 mg of sample) after each measurement. This normalized integrated absorbance is commonly used in SS-ETAAS to compare signals, as it is practically impossible to introduce a sample mass of exactly 1.00 mg, or always to introduce exactly the same sample mass in a series of measurements. Liquid calibration or modifier solutions were pipetted manually onto the platform using a microlitre pipette.
Typical graphite furnace temperature programs used under the various conditions are given in Tables 1a–d . Alterations of these programs used in several experiments are given in the Results and discussion section. The drying stage was used only when aqueous calibration solutions were analysed or when a modifier solution was pipetted on top of the solid sample. In all other cases the drying stage was omitted. Argon of 99.996% purity (White Martins, São Paulo, Brazil) was used as the purge and protective gas. For some experiments the solid sampling platform was treated with 400 µg of ruthenium as a permanent modifier by pipetting 10 aliquots of 40 µL each of a 1000 mg L−1 ruthenium stock solution, and executing the temperature program shown in Table 2 after each injection. One such treatment with ruthenium was sufficient for the entire lifetime of a platform.
| (a) | ||||
|---|---|---|---|---|
| Program stage | Temperature/°C | Ramp/°C s−1 | Hold time/s | Gas flow rate/L min−1 |
| 1 Pyrolysis | 800 | 100 | 20 | 2.0 |
| 2 Auto zero | 800 | 0 | 6 | Stop |
| 3 Atomizationa | 1700 | 2000 | 4 | Stop |
| 4 Cleaning | 2300 | 100 | 3 | 2.0 |
| (b) | ||||
|---|---|---|---|---|
| Program stage | Temperature/°C | Ramp/°C s−1 | Hold time/s | Gas flow rate/L min−1 |
| 1 Drying | 110 | 10 | 20 | 2.0 |
| 2 Drying | 140 | 10 | 30 | 2.0 |
| 3 Pyrolysis | 700 | 100 | 20 | 2.0 |
| 4 Auto zero | 700 | 0 | 4 | Stop |
| 5 Atomizationa | 2400 | 2000 | 5 | Stop |
| 6 Cleaning | 2400 | 0 | 3 | 2.0 |
| (c) | ||||
|---|---|---|---|---|
| Program stage | Temperature/°C | Ramp/°C s−1 | Hold time/s | Gas flow rate/L min−1 |
| 1 Pyrolysis | 350 | 100 | 10 | 2.0 |
| 2 Auto zero | 350 | 0 | 6 | Stop |
| 3 Atomizationa | 1950 | 1500 | 5 | Stop |
| 4 Cleaning | 2400 | 1000 | 5 | 0.1 |
| (d) | ||||
|---|---|---|---|---|
| Program stage | Temperature/°C | Ramp/°C s−1 | Hold time/s | Gas flow rate/L min−1 |
| a Integrated absorbance signal recorded in this stage.b For solid sampling platforms treated with ruthenium.c For untreated pyrolytic graphite-coated solid sampling platforms. | ||||
| 1 Drying | 195 | 15 | 35 | 2.0 |
| 2 Pyrolysis | 350 | 50 | 15 | 0.1 |
| 3 Pyrolysis | 450b/800c | 25 | 20 | 0.1 |
| 4 Auto zero | 450b/800c | 0 | 6 | Stop |
| 5 Atomizationa | 1950 | 1500 | 5 | Stop |
| 6 Cleaning | 2400 | 1000 | 5 | 0.1 |
| Program stage | Temperature/°C | Ramp/°C s−1 | Hold time/s | Gas flow rate/L min−1 |
|---|---|---|---|---|
| 1 Drying | 100 | 7 | 10 | 2.0 |
| 2 Drying | 130 | 7 | 40 | 2.0 |
| 3 Drying | 160 | 100 | 60 | 2.0 |
| 4 Pyrolysis | 1000 | 100 | 20 | 2.0 |
| 5 Pyrolysis | 1400 | 100 | 5 | 0.1 |
| 6 Pyrolysis | 2000 | 100 | 5 | 0.1 |
Reagents and reference materials
Analytical grade reagents were used throughout. The nitric acid (Merck) used to prepare the aqueous calibration standards was further purified by sub-boiling distillation in a quartz sub-boiling still (Kürner Analysentechnik, Rosenheim, Germany). All containers and glassware were soaked in 3 mol L−1 nitric acid for at least 24 h and rinsed three times with deionized water before use. Distilled, de-ionized water with a specific resistivity of 18 MΩ cm from a Milli-Q water purification system (Millipore, Bedford, MA, USA) was used for the preparation of calibration and modifier solutions.The thallium stock standard solution, 1000 mg L−1, was prepared from thallium nitrate (Merck) in 0.5 mol L−1 HNO3. The working standards were prepared by serial dilutions of the stock solution in 0.014 mol L−1 nitric acid.
The following solutions were used as chemical modifiers in the different experiments: 1000 mg L−1 Ru in 8% HCl (Merck) for treating the solid sampling platforms with ruthenium as a permanent modifier, using the program in Table 2; 1000 mg L−1 Pd as the nitrate (Merck); and 100 g L−1 NH4NO3 (Merck) in 0.05% (v/v) Triton X-100 (Union Carbide).
The following reference materials were used in this work: CRM 320 Trace Elements in River Sediment (Community Bureau of Reference, Brussels, Belgium); SRM 2704 Buffalo River Sediment and SRM 1646a Estuarine Sediment (National Institute of Standards and Technology, Gaithersburg, MD, USA); BCSS-1, HISS-1, MESS-1, MESS-2, MESS-3 and PACS-2 Marine Sediments (National Research Council Canada, Ottawa, Canada); and RS-1, RS-2, RS-3 and RS-4 river sediment samples that had been previously analysed in a round robin test, and for which mean values and a 95% confidence interval for a large number of elements have been published.37 In most experiments the sediment samples and reference materials were used as supplied without further grinding. According to the certificates, the NRCC marine sediment reference materials (BCSS-1, HISS-1, MESS-1, MESS-2, MESS-3 and PACS-2) were ‘screened to pass a No. 120 (125 µm) screen’; the NIST SRM 2704 Buffalo River Sediment was ‘screened and passed through a 100 mesh sieve and retained on a 400 mesh sieve’, i.e., has a particle size distribution of 38–150 µm; and the NIST SRM 1646a Estuarine Sediment was ‘ball-milled to pass a sieve with openings of 150 µm (No. 100)’. A particle size analysis was carried out for river sediments R 1–4 using laser diffraction, and ‘the fraction >100 μm was found to be less than 5%’. No information on particle size distribution was available for CRM 320. For some experiments sediment samples were further ground in an agate mortar to a particle size ≤50 µm, however, the results were no different from those without grinding and are not discussed here.
Results and discussion
Analysis of river sediments
The NIST SRM 2704 Buffalo River Sediment was chosen for all optimization and calibration purposes, as it was the only sediment reference material with a certified value for thallium (1.06 ± 0.07 µg g−1) that was available in our laboratories at that time. First we had to decide between the use of standard graphite tubes with a dosing hole, or of solid sampling graphite tubes without a dosing hole. The latter ones were chosen for all future experiments because the sensitivity (in integrated absorbance) with these tubes was 25% higher than with the standard tubes, as there was no loss of analyte atoms by diffusion through the dosing hole. The only disadvantage was that any aqueous calibration or modifier solution had to be pipetted manually onto the solid sampling platforms and introduced into the graphite tube in the same way as a solid sample.The pyrolysis curves for 1.0 ng Tl in dilute nitric acid solution (10 µL of a 100 µg L−1 standard) and for the Buffalo River Sediment reference material, normalized to the same mass of 1.0 ng Tl, without the use of any modifier, are shown in Fig. 1. It is obvious that, except for a minor difference in sensitivity, the analyte exhibited exactly the same behavior in aqueous solution and in the solid sediment sample. A pyrolysis temperature of 800°C could be used without the risk of analyte losses even without the addition of a modifier. At an atomization temperature of 1700°C, the Tl signal had a half width of about 0.8 s and a total width at the baseline of about 2 s with a minimum of tailing, and the background absorption under these conditions was negligible. Hence, these conditions, which are summarized in Table 1a, were adopted
for all future experiments with river sediments.
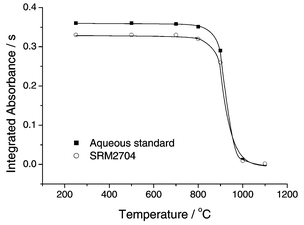 | ||
| Fig. 1 Pyrolysis curves for 1 ng Tl in dilute nitric acid solution and in NIST SRM 2704 Buffalo River Sediment without the use of a chemical modifier. | ||
The calibration curve for aqueous thallium standards, using the average of three measurements each of standard solutions containing 0.00, 0.25, 0.50, 0.75 and 1.00 ng Tl, was linear at least to an integrated absorbance of 0.4 s, following the linear regression equation Aint = 0.0042 + 0.368 m (R = 0.9988). For the calibration curve established by weighing 10 different arbitrary masses between 0.1 and 1 mg of the solid reference material SRM 2704, a quadratic relationship Aint = 0.00097 + 0.352 m − 0.0137 m2 (R = 0.9990) gave a slightly better correlation. This is in agreement with previous experience,36 and results from the fact that there is typically one order of magnitude difference in the amount of sediment introduced into the graphite furnace between the lowest and the highest calibration point, when only one reference material is used for calibration. The characteristic mass resulting from the slope of the calibration curves was m0 = 13.0 pg Tl, a value that is close enough to the theoretical value of 9.9 pg calculated by L'vov,38 taking into account the slightly different tube dimensions. The results of the thallium determination in the other river sediment reference materials, using direct solid sampling and both correlation equations for calibration, are shown in Table 3. All values obtained with calibration against the solid SRM and using aqueous calibration standards are in excellent agreement with the mean values and well within the 95% confidence interval of the round robin for RS-1–RS-4, or the non-certified concentration in the BCR-320 sediment. A limit of detection of 0.02 µg g−1 Tl was determined using the ‘zero-mass response’ as proposed by Kurfürst,39i.e., by using the integrated absorbance of empty solid sample platforms inserted repeatedly (n = 10) into the furnace, followed by the usual temperature program, as the measure of the blank value.
| Reference material | Reference value/µg g−1 | Solid calibrationa/µg g−1 | Aqueous calibrationa/µg g−1 |
|---|---|---|---|
| a Average and standard deviation of five independent determinations.b Mean value and 95% confidence interval of round robin from Ref. 37.c Non-certified concentration. | |||
| RS–1 | 1.12 ± 0.14b | 1.18 ± 0.12 | 1.13 ± 0.12 |
| RS–2 | 0.48 ± 0.08b | 0.46 ± 0.04 | 0.44 ± 0.04 |
| RS–3 | 0.68 ± 0.10b | 0.67 ± 0.05 | 0.64 ± 0.04 |
| RS–4 | 0.35 ± 0.06b | 0.35 ± 0.03 | 0.34 ± 0.03 |
| BCR-320 | (0.5)c | 0.48 ± 0.03 | 0.46 ± 0.03 |
Analysis of marine sediments using continuum-source background correction
When the temperature program of Table 1a was applied for the analysis of marine sediment samples, very erratic signals for thallium were obtained and significant background absorption was observed, calling for a modification of the temperature program and the use of a chemical modifier. In the first attempt we were following the recommendations of Welz et al.16 and tried to use reduced palladium as a modifier by introducing a palladium solution into the solid sampling platform, and heating it to 1000°C before the solid sample was weighed onto the platform. The introduction of 5 µg of palladium did not bring about any significant improvement, hence we increased the mass of palladium up to 100 µg and began to analyse the three marine sediment reference materials, MESS-2, PACS-2 and HISS-1, with a non-certified thallium content of 0.98 µg g−1, 0.6 µg g−1
and 0.06 µg g−1, respectively.Using a pyrolysis temperature of 700°C and an atomization temperature of 1800°C, the background signal was reasonably low, which is a prerequisite for the application of continuum-source background correction. However, the results we obtained for the three sediments were not encouraging. A closer look at the peak shapes, particularly that for HISS-1 which is shown in Fig. 2a, clearly exhibited a spectral interference that, in spite of the low background absorption, could not be corrected by the continuum-source background corrector. The extent of this interference was different for the different sediment samples and/or there was more than one interfering species, the concentrations and ratios of which were different, depending on the matrix composition of the sediment.
 | ||
| Fig. 2 Absorption signals for thallium (solid line) and background (broken line) for marine sediment reference materials using reduced palladium as a chemical modifier and continuum-source background correction: (a) HISS-1, 700°C pyrolysis and 1800°C atomization temperature; (b) MESS-2, 700°C pyrolysis and 2200°C atomization temperature; (c) HISS-1, 700°C pyrolysis and 2200°C atomization temperature; (d) blank signal from 100 µg pre-reduced palladium, 700°C pyrolysis and 2200°C atomization temperature. | ||
In an attempt to improve the situation we increased the atomization temperature to 2200°C and we investigated the use of hydrogen as an alternate gas, which, however, did not change the situation significantly. The results obtained for MESS-2 were around 0.6 µg g−1 Tl, which was obviously, at least in part, due to the over-correction of the spectral background that became apparent (Fig. 2b). For HISS-1 the sign of the interference changed with increasing background at the higher atomization temperature, as is shown in Fig. 2c, and the integrated absorbance values became negative. The early, sharp background signal was at least in part due to a spectral interference from palladium, which is described in the literature,40,41 and which becomes apparent in the blank signal in Fig. 2d.
This interference might also have contributed to the distortion of the early part of the thallium signal in Figs. 2b and 2c. After this negative experience we discontinued the use of palladium as a modifier for this type of sample.
The ammonium nitrate modifier
First we consulted the certificates for the above reference samples and several other marine sediment reference materials for information about potentially interfering major constituents. There is roughly an order of magnitude difference in the concentrations of iron, sulfur and chloride in the various marine sediments, which may well explain some of the observed effects. However, the most obvious difference is in the chloride content of marine sediments, which is at the percentage level, and 2-3 orders of magnitude higher compared to the river sediments. Although chloride cannot be responsible for all the interference effects observed with marine sediments, it was considered an important first step to look for a modifier that could remove most of this matrix element, and ammonium nitrate was an obvious choice for that purpose. This modifier was first proposed by Ediger et al.42 in 1974, and has also been used successfully together with palladium to overcome chloride interferences in the determination of thallium.17We investigated the use of the ammonium nitrate modifier in three different forms: i) ammonium nitrate alone, added in solution on top of the solid sample on the platform; ii) same as i), but the platform was treated with ‘reduced palladium’ before the solid sample was weighed onto the platform; and iii) same as i), but the platform was treated with ruthenium as a permanent modifier according to the program in Table 2 before the solid sample was weighed onto the platform. The advantage of iii) over ii) was that the treatment with ruthenium had to be done only once for the lifetime of the platform, whereas the treatment with palladium had to be repeated after each atomization cycle, making the latter procedure relatively slow and expensive.
The temperature program in Table 1b is the result of a careful optimization and was used for all three conditions. There was still a high background absorption with all three modifier combinations and there was some baseline distortion associated with this background, as can be seen in Fig. 3a for MESS-2 with the ammonium nitrate modifier. However, the signals were clearly separated in time so that the background absorption and the baseline distortion (which would have affected the integrated absorbance signal) could be excluded from the measurement by proper timing of the integration window, as is shown in Fig. 3b. The results obtained under these conditions, and by calibration against SRM 2704 Buffalo River Sediment using the same modifiers, are summarized in Table 4. It is obvious that the results are significantly better than the earlier ones, but with a few exceptions they are not yet satisfactory. There was no clear trend in favor of one of the modifiers. For two sediments, HISS-1 and SRM 1646a, the ammonium nitrate alone as a modifier gave the highest recovery of thallium. For three sediments, MESS-1, MESS-2 and MESS-3, the combination of ammonium nitrate with reduced palladium gave the highest values; for BCSS-1, ammonium nitrate with and without reduced palladium gave the same values; and for PACS-2, ammonium nitrate with palladium or ruthenium gave essentially the same result which was about a factor of two higher than that with ammonium nitrate alone.
 | ||
| Fig. 3 Absorption signals for thallium (solid line) and background (broken line) for MESS-2 with 1 mg of ammonium nitrate as the modifier using the temperature program in Table 1(b) and continuum-source background correction: (a) 6 s measurement time; (b) 1.5 s measurement time. | ||
| Reference material | Reference value | NH4NO3a | Pd/NH4NO3b | Ru/NH4NO3c |
|---|---|---|---|---|
| a 10 µL of 10% (w/v) NH4NO3.b 5 µg Pd reduced + 10 µL of 10% (w/v) NH4NO3.c 400 µg Ru (permanent modifier) + 10 µL of 10% (w/v) NH4NO3.d Non-certified concentration.e Certified value and 95% confidence interval. | ||||
| MESS–1 | 0.7d | 0.40 ± 0.05 | 0.47 ± 0.05 | 0.42 ± 0.05 |
| MESS–2 | 0.98d | 0.86 ± 0.06 | 0.92 ± 0.07 | 0.87 ± 0.03 |
| MESS–3 | 0.9 ± 0.06e | 0.83 ± 0.05 | 0.92 ± 0.10 | 0.85 ± 0.10 |
| PACS–2 | 0.6d | 0.11 ± 0.07 | 0.20 ± 0.10 | 0.21 ± 0.06 |
| BCSS–1 | 0.6d | 0.43 ± 0.04 | 0.43 ± 0.02 | 0.36 ± 0.03 |
| HISS–1 | 0.06d | 0.06 ± 0.02 | 0.04 ± 0.01 | 0.04 ± 0.01 |
| SRM 1646a | <0.5d | 0.16 ± 0.02 | 0.14 ± 0.02 | 0.10 ± 0.02 |
Although there is no trend in the results obtained in this set of experiments that would favor one of the modifiers, there is an interesting correlation between the recovery of thallium and the sulfur content of the sediments, which is outlined in Table 5. HISS-1 has not been included in this table as there is no information available about its sulfur content, and for SRM 1646a, a thallium content of 0.2 µg g−1 was used instead of <0.5 µg g−1 that is given in the certificate, as we have good reasons to believe that this is the true content.43 Although this might be a gross simplification, the average value for thallium found with all three modifiers was taken for comparison, but the trend in Table 5 is obvious. The marine sediment reference materials are arranged according to their decreasing sulfur content, and the recovery of thallium shows a proportionally increasing tendency. When the thallium signal suppression (the inverse of the recovery in Table 5) was plotted as a function of the sulfur content, a linear regression equation of y = 3.1434 + 54.1585x was obtained with a correlation coefficient of R = 0.948, and a probability factor of p = 0.00115, exhibiting a high level of significance. Hence, it could be assumed that sulfur is the cause of the low thallium recoveries, particularly as there are reports in the literature about a sulfate interference on thallium.22 The fact that Zendelovska and Stafilov23 found that it was not possible to determine thallium directly from solutions of sulfide minerals may be due to the same interference. There are good reasons to believe that this interference is of spectral origin, as Welz et al.,44 upon vaporizing magnesium sulfate in a graphite furnace, measured a molecular spectrum with a rotational fine structure between 250 nm and 310 nm and a maximum around 280 nm that they interpreted as the spectrum of the SO2 molecule. The fact that Welz et al.45 could not find any significant influence from up to 1 g L−1 of sulfate on the determination of thallium in an instrument with Zeeman-effect background correction further supports the assumption that this interference is of spectral origin.
| Reference material | Sulfur content (%) | Tl content/µg g−1 | Tl found/µg g−1 | Recovery (%) |
|---|---|---|---|---|
| a Non-certified concentration.b Best estimate according to independently obtained results.43c Certified value and 95% confidence interval. | ||||
| PACS–2 | 1.29 | 0.6a | 0.17 | 29 |
| MESS–1 | 0.72 | 0.7a | 0.43 | 61 |
| BCSS–1 | 0.36 | 0.6a | 0.41 | 68 |
| SRM 1646a | 0.352 | 0.2b | 0.13 | 67 |
| MESS–3 | 0.19 | 0.9 ± 0.06c | 0.87 | 97 |
| MESS–2 | 0.18 | 0.98a | 0.88 | 90 |
Measurements with Zeeman-effect background correction
As we suspected that a spectral interference due to a molecular absorption spectrum with a rotational fine structure, which cannot be corrected by continuum-source background correction, in addition to the well-known chloride interference was causing the problems encountered in this work, we repeated some of the measurements using Zeeman-effect background correction. Firstly, we investigated if it was possible to omit the pyrolysis stage when Zeeman-effect background correction was used, as proposed by Manning and Slavin,19 and just use a ‘modified drying stage’ at 350°C, as reported by López-García et al.,20 for the determination of thallium in soils and sediments (temperature program of Table 1c). Secondly, we used ammonium nitrate as a chemical modifier, added in solution on top of the solid samples on the platform, and a pyrolysis temperature of 800°C
in order to remove the majority of the chloride prior to the atomization stage (temperature program of Table 1d). Thirdly, we treated the solid sampling platform with ruthenium as a permanent modifier (temperature program of Table 2) and then added ammonium nitrate in solution on top of the solid samples on the platform, as in the previous experiment. Calibration against a working curve established with different masses of SRM 2704 (Buffalo River Sediment), treated in the same way as the marine sediment samples, was used throughout.The results of these experiments are summarized in Table 6, and some typical atomic and background signals obtained under the different conditions are shown in Fig. 4. It is obvious that the results without a modifier are far too low. It is also obvious from the signals in Fig. 4 that a relatively high background signal is observed, which is according to expectation under these conditions. However, there is no distortion of the baseline or the atomization signals, which means that the background correction is working properly, and there is no spectral interference that could be responsible for the low results. As TlCl is volatile only above 400°C, analyte losses in the pyrolysis stage at 350°C can be excluded as well. This means that the low recovery of thallium from these samples is most likely due to volatilization of the monochloride in the atomization
stage, which can be atomized only in part due to its relatively high dissociation energy of 88 ± 0.5 kcal mol−1.5
 | ||
| Fig. 4 Absorption signals for thallium (solid line) and background (broken line) for HISS-1, PACS-2 and MESS-2 marine sediment reference materials, using Zeeman-effect background correction, without a modifier, with 1 mg of ammonium nitrate as the modifier (NH4NO3), and with 400 µg ruthenium as a permanent modifier and 1 mg of ammonium nitrate added on top (Ru + NH4NO3). | ||
| Reference material | Reference value | ICP-MSa | Found value/µg g−1 | ||
|---|---|---|---|---|---|
| Withoutb | NH4NO3c | Ru + NH4NO3d | |||
| a ETV-ICP-MS with isotopic dilution calibration.21b Without chemical modifier; temperature program as in Table 1(c).c With 10 µL of 10% NH4NO3 added in solution on top of the solid sample; temperature program as in Table 1(d).d With 400 µg Ru as permanent modifier and 10 µL of 10% NH4NO3 added in solution on top of the solid sample; temperature program as in Table 1(d).e Information value.f Certified value and 95% confidence interval. | |||||
| HISS–1 | (0.06)e | 0.055 ± 0.004 | 0.02 ± 0.002 | 0.03 ± 0.003 | 0.05 ± 0.003 |
| PACS–2 | (0.6)e | 0.52 ± 0.02 | 0.12 ± 0.03 | 0.22 ± 0.04 | 0.58 ± 0.02 |
| MESS–2 | (0.98)e | 0.99 ± 0.04 | 0.19 ± 0.01 | 0.72 ± 0.01 | 1.07 ± 0.03 |
| MESS–1 | (0.7)e | 0.70 ± 0.02 | |||
| MESS–3 | 0.90 ± 0.06f | 1.08 ± 0.07 | |||
| BCSS–1 | (0.6)e | 0.59 ± 0.04 | |||
| SRM1646a | (<0.5)e | 0.20 ± 0.02 | |||
The addition of ammonium nitrate as a chemical modifier and the increase in the pyrolysis temperature markedly reduced the background absorption (Fig. 4), which is again according to expectation, as is the increase in the recovery of thallium (Table 6). However, these values are still below expectation. It would require additional experiments to find out if these analyte losses occur during the pyrolysis or the atomization stage, but the results indicate that ammonium nitrate alone cannot prevent the interference completely. Only with the additional stabilizing power of ruthenium were the results for thallium within the expected range, as can be seen in Table 6.
This is the second report on the application of ruthenium as a permanent chemical modifier for the direct analysis of solid samples, after it had already been applied successfully for the determination of cadmium and lead in mineral coal.36 We have good reasons to believe that the low value of 0.20 µg g−1 Tl for SRM 1646a (non-certified reference value < 0.5 µg g−1) is close to the true value, as a very similar result has been obtained using an independent technique.43
Conclusion
The determination of thallium in river sediments can be carried out easily with direct solid sampling using continuum-source background correction with ETAAS and calibration against solid sediment reference materials or against a working curve established with aqueous standards. The determination of thallium in marine sediments required the use of Zeeman-effect background correction and the application of chemical modifiers. The combination of a platform treatment with ruthenium as a permanent modifier and the addition of ammonium nitrate in solution on top of the solid sample provided the best results. This additional example confirms that it is possible to use permanent chemical modifiers for the direct analysis of solid samples, which greatly facilitates and simplifies the use of this technique for routine purposes. As a result of the experience with river sediments, where calibration against aqueous standards gave essentially the same results as calibration against SRM 2704, it may be assumed that aqueous standards may be also used for calibration for marine sediments under optimized conditions instead of SRM 2704. This alternative, however, has not been evaluated experimentally.Acknowledgements
The authors wish to thank Aline Klassen and Isabel Cristina S. Ferreira for their careful execution of part of the experiments. The authors are also grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for a research scholarship awarded to B.W., to Fundação de Amparo e Pesquisa do Rio Grande do Sul (FAPERGS) for financial support, and to Analytik Jena AG for the loan of an atomic absorption spectrometer.References
- J. Falbe and M. Regitz, Römpp Chemie Lexikon, Thieme, Stuttgart, New York, 1992, 9th edn., vol. 6, p. 4549 Search PubMed.
- K. Bunzl, M. Trautmannsheimer, P. Schramel and W. Reifenhauser, J. Environ. Qual., 2001, 30, 934 Search PubMed.
- M. S. Leloux, N. P. Lich and J. R. Claude, At. Spectrosc., 1987, 8, 71 Search PubMed.
- W. Slavin, D. C. Manning and G. R. Carnrick, At. Spectrosc., 1981, 2, 137 Search PubMed.
- B. V. L'vov, Spectrochim. Acta, Part B, 1978, 33, 153 CrossRef.
- T. M. Mahmood and K. W. Jackson, Spectrochim. Acta, Part B, 1996, 51, 1155 CrossRef.
- G. Chen and K. W. Jackson, Spectrochim. Acta, Part B, 1998, 53, 981 CrossRef.
- E. Bulska and H. M. Ortner, Spectrochim. Acta, Part B, 2000, 55, 491 CrossRef.
- W. Slavin and D. C. Manning, Spectrochim. Acta, Part B, 1980, 35, 701 CrossRef.
- W. Slavin, G. R. Carnrick and D. C. Manning, Anal. Chim. Acta, 1982, 138, 103 CrossRef CAS.
- W. Slavin, G. R. Carnrick and D. C. Manning, Anal. Chem., 1984, 56, 163 CrossRef CAS.
- B. Welz, G. Schlemmer and J. R. Mudakavi, J. Anal. At. Spectrom., 1988, 3, 695 RSC.
- X-q. Shan, Z-m. Ni and L. Zhang, Talanta, 1984, 31, 150 CrossRef CAS.
- B. Welz, G. Schlemmer and J. R. Mudakavi, J. Anal. At. Spectrom., 1988, 3, 93 RSC.
- L. M. Voth-Beach and D. E. Shrader, J. Anal. At. Spectrom., 1987, 2, 45 RSC.
- B. Welz, G. Schlemmer and J. R. Mudakavi, Anal. Chem., 1988, 60, 2567 CrossRef CAS.
- Q. Yang and J. Smeyers-Verbeke, Clin. Chim. Acta, 1991, 204, 23 CrossRef CAS.
- X-f. Yin, G. Schlemmer and B. Welz, Anal. Chem., 1987, 59, 1462 CrossRef CAS.
- D. C. Manning and W. Slavin, Spectrochim. Acta, Part B, 1988, 43, 1157 CrossRef.
- I. López-García, M. Sánchez-Merlos and M. Hernández Córdoba, Anal. Chim. Acta, 1996, 328, 19 CrossRef CAS.
- S. M. Maia, M. G. R. Vale, B. Welz and A. J. Curtius, Spectrochim. Acta, Part B, 2001, 56, 1263 CrossRef.
- K. Matsusaki and T. Oishi, Anal. Sci., 1993, 9, 381 Search PubMed.
- D. Zendelovska and T. Stafilov, Anal. Sci., 2001, 17, 425 Search PubMed.
- P. J. Potts, Handbook of Silicate Rock Analysis, Blackie, Glasgow, 1987 Search PubMed.
- A. A. Verbeek, M. C. Mitchell and A. M. Ure, Anal. Chim. Acta, 1982, 135, 215 CrossRef CAS.
- M. Bettinelli, Anal. Chim. Acta, 1983, 148, 193 CrossRef CAS.
- D. C. Bartenfelder and A. D. Karathanasis, Commun. Soil Sci. Plant Anal., 1988, 19, 471 Search PubMed.
- H. Agemian and E. Bedek, Anal. Chim. Acta, 1980, 119, 323 CrossRef CAS.
- J. W. McLaren, D. Beauchemin and S. S. Berman, Anal. Chem., 1987, 59, 610 CrossRef CAS.
- B. Welz and M. Sperling , Atomic Absorption Spectrometry, Wiley-VCH, Weinheim, 1999, 3rd edn., pp. 682–683 Search PubMed.
- E. Macalalad, R. Bayoran, B. Ebarvia and I. Rubeška, J. Geochem. Explor., 1988, 30, 167 CrossRef CAS.
- E. Ivanova, G. Gentscheva, M. Stoimenova and I. Havezov, Anal. Lab., 1995, 4, 14 Search PubMed.
- T.-z. Guo and J. Baasner, J. Autom. Chem., 1996, 18, 221 Search PubMed.
- E. Waidmann, M. Stoeppler and P. Heininger, Analyst, 1992, 117, 295 RSC.
- J. B. B. da Silva, M. A. M. da Silva, A. J. Curtius and B. Welz, J. Anal. At. Spectrom., 1999, 14, 1737 RSC.
- M. G. R. Vale, M. M. Silva, B. Welz and É. C. Lima, Spectrochim. Acta, Part B, 2001, 56, 1859 CrossRef.
- P. Heininger, J. Pelzer, R. Henrion and G. Henrion, Fresenius' J. Anal. Chem., 1998, 360, 344 CrossRef CAS.
- B. V. L'vov, Spectrochim. Acta, Part B, 1990, 45, 633 CrossRef.
- U. Kurfürst, Solid sample analysis, Springer, Berlin, Heidelberg, New York, 1998, p. 115 Search PubMed.
- F. Vajda, Anal. Chim. Acta, 1981, 128, 31 CrossRef CAS.
- W. Slavin and G. R. Carnrick, At. Spectrosc., 1986, 7, 9 Search PubMed.
- R. D. Ediger, G. E. Peterson and J. D. Kerber, At. Absorpt. Newsl., 1974, 13, 61 Search PubMed.
- B. Welz, M. G. R. Vale, M. M. Silva, H. Becker-Ross, M-d. Huang, S. Florekand U. Heitmann, Spectrochim. Acta, Part B, submitted for publication Search PubMed.
- B. Welz, G. Bozsai, M. Sperling and B. Radziuk, J. Anal. At. Spectrom., 1992, 7, 505 RSC.
- B. Welz, G. Schlemmer and J. R. Mudakavi, J. Anal. At. Spectrom., 1992, 7, 1257 RSC.
| This journal is © The Royal Society of Chemistry 2002 |