Continuous flow chemical vapour generation of silver for atomic absorption spectrometry using tetrahydroborate(III) reduction—system performance and assessment of the efficiency using instrumental neutron activation analysis
Tomáš
Matoušek
*,
Jiří
Dědina
and
Miloslav
Vobecký
Academy of Sciences of the Czech Republic, Institute of Analytical Chemistry, Laboratory of Trace Element Analysis, Vídeňská 1083, CZ-142 20, Prague, Czech Republic. E-mail: matousek@biomed.cas.cz
First published on 5th December 2001
Abstract
The chemical vapour generation (CVG) of silver by tetrahydroborate(III) reduction as a sample introduction technique for atomic absorption spectrometry was studied. 7% of the analyte was volatilized in a miniature batch arrangement, as determined by instrumental neutron activation analysis. The continuous flow silver CVG system was designed to be interfaced with a diffusion flame or multiple microflame quartz tube atomizer, and peak area characteristic masses of 7 and 0.6 ng, respectively, were achieved. The chemical vapour generation efficiency of the described continuous flow system was in the same range as that of the batch system.
Introduction
The most important reaction for analyte volatilization as a sample introduction technique for analytical atomic spectroscopy is analyte reduction by tetrahydroborate(III) ions in acidic media at room temperature. Except for the Group III–VI elements, traditionally volatilized as hydrides1 or mercury cold vapours, the chemical generation of volatile species (possibly as hydrides, vapours or other unidentified species) of noble and transition metals has recently been reported. Besides the relatively well established volatilization of cadmium,2–5 reports on copper,4–7 nickel,5,8 zinc,4 silver,4,7 gold,4,7 iron and cobalt,5 and rhodium and palladium7 have appeared.Luna et al.4 described a miniature batch system for the CVG of Ag, Au, Cd, Cu and Zn by tetrahydroborate(III) with a heated quartz tube atomizer and an AAS detector. The assessed efficiency of the analyte release (i.e., the generation and gas–liquid separation1) was 92 ± 4, 80–95, 85–90 and 87% for Au, Ag, Cu and Zn, respectively, based on inductively coupled plasma mass spectrometry (ICP-MS) measurements of residual analytes in solution after the CVG process. This was in a sharp contrast with overall efficiencies estimated from the relative comparison of sensitivities with those reported for flame AAS, which were 0.6, 8, 4 and 75% for Au, Ag, Cu and Zn, respectively.4 The discrepancy was explained by the transport losses of the volatilized analyte on the way to the atomizer and/or the poor atomizer performance for these elements.4 Sturgeon et al.6 reported the removal of 50% Cu from solution after the NaBH4 reaction in a batch arrangement. Other literature references give low CVG efficiencies (i.e., efficiency of release and transport to the detector): Guo et al.8 achieved the same Ni signal intensity for nebulization and volatile species generation using ICP- OES; and Pohl and Zyrnicki5 reported maximum CVG efficiencies of 2.3% and 1.4% for Cu and Cd, respectively, and significant but low (<0.1%) efficiencies for Co, Fe and Ni.
The only approach to the CVG of transition and noble metals for AAS described in the literature4 employed the batch generation method, which is inferior to flow methods because the batch mode is difficult to automate and requires much more manual input on the part of the operator.1 On the other hand, the continuous flow systems described in the literature5–8 were interfaced to an ICP torch and were not optimized for the low purge gas flow rates that are required for sensitive AAS (and/or possibly AFS) detection.
The aim of our work was to develop a continuous flow (CF) system to volatilize transition and noble metals, applicable for AAS and AFS detection. Ag was chosen as the model analyte. To appraise the performance of the atomization units, initial testing was performed using a batch CVG system analogous to that of ref. 4. To provide an accurate estimate of the efficiency of the silver release in the batch system, the residual analyte was determined after the volatilization process. Instrumental neutron activation analysis (INAA) was the method of choice, as it allows the determination of sample residues in situ, regardless of the chemical form and without any subsequent sample treatment.
Experimental
Chemicals
All reagents were of analytical grade or better. Doubly distilled water was used throughout.Ag standard solutions were prepared by dilution of 1000 µg ml−1 stock solution (BDH, UK) with 1 M HNO3. The reductant, 1% NaBH4 in 0.2% KOH (m/v), was prepared fresh daily. It was filtered through a paper filter (Filtrak 389, Barenstein, Germany) at atmospheric pressure before use.
AAS measurements
A Model 503 AAS spectrometer with a Model 56 strip chart recorder (Perkin-Elmer, Norwalk, CT, USA) with no background correction was used. The light source was a Ag hollow cathode lamp (Perkin-Elmer) operated at 12 mA, at wavelength 328 nm and bandwidth 0.7 nm.Chemical vapour generation systems
 | ||
| Fig. 1 Continuous flow system with the diffusion flame atomizer, with details of the capillary tips section. | ||
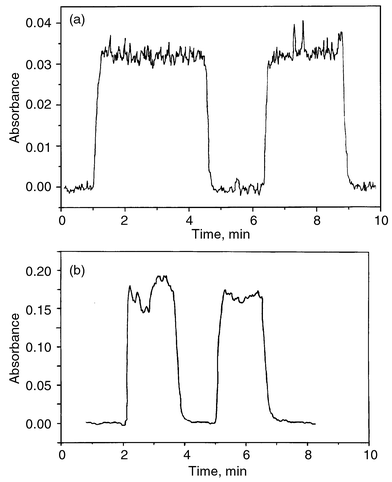 | ||
| Fig. 2 (a) Continuous flow-CVG-diffusion flame signal; Ag, 5 µg ml−1 and time constant, 0.5 s. (b) Continuous flow-CVG-MMQTA signal; Ag, 1 µg ml−1 and time constant, 2 s. | ||
Atomizers
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C by a laboratory-made electrical furnace (120 mm
heated length, 15 mm id) controlled by a Rex C-100 controller (SYSCON-RKC, IN, USA) with a K-type thermocouple sensor. 50 ml min−1 of argon or 100 ml min−1 of 10% H2 in Ar was used as a purge gas, with this atomizer connected to the CF or batch system, respectively. The air flow at a rate that provided optimum oxygen intake (1 ∶ 3 O2 to H2 ratio9) served as an outer gas.
°C by a laboratory-made electrical furnace (120 mm
heated length, 15 mm id) controlled by a Rex C-100 controller (SYSCON-RKC, IN, USA) with a K-type thermocouple sensor. 50 ml min−1 of argon or 100 ml min−1 of 10% H2 in Ar was used as a purge gas, with this atomizer connected to the CF or batch system, respectively. The air flow at a rate that provided optimum oxygen intake (1 ∶ 3 O2 to H2 ratio9) served as an outer gas.
The MMQTA was connected to the generation systems using PTFE transfer tubing (150 mm long, 1 mm id).
INAA measurements
The modified batch generator was used for the preparation of samples.10 µl of blank or Ag standard solution were pipetted onto a circular polyethylene foil target (0.25 mm thick, 22 mm diameter), which replaced the lower (pipe-shaped) part of the batch system. 32 µl of a treatment solution (sodium tetrahydroborate(III) or sodium hydrogencarbonate, see Table 1) were then added in the same manner as in the batch generation system, using the upper part of the batch system (see above). After the reaction, the droplets of liquid were soaked into a filter paper circle (10 mm diameter) and left on the polyethylene target to dry freely for two days. The dried samples were covered by another polyethylene circle, which was fused to the bottom one. The targets were then wrapped in aluminium foil and irradiated with thermal neutrons in the core of an experimental light water nuclear reactor (LWR15, Nuclear Research Institute, Řežu Prahy, Czech Republic) at a thermal neutron flux of 1.3 × 1014 cm−2 s−1 for 2 hours. The measurements of the 110mAg isotope (half-life = 249.9 days) were started 26 days after irradiation, using an α-spectrometer (Canberra, USA) with a high purity germanium detector (20% relative efficiency). Peak area measurements of the intense 675.7 keV and 884.7 keV 110mAg α-lines were used for quantification with a measurement time of 200 000 s. The results obtained at both lines differed by less than 0.5%.| Samplea | Treatmenta | Ag found (%) | Remarks | |
|---|---|---|---|---|
| a Blank, 10 µl of 1 M HNO3; Ag, 100 ng Ag in 10 µl of 1 M HNO3; BH4−, 32 µl of 1% w/w NaBH4 in 0.2% KOH; CO32−, 32 µl of 8.5% NaHCO3 + 2.5% Na2B4O7 in 0.2% KOH (w/w). | ||||
| i | Blank | BH4− + Ag after 10 min delay | Reference—100.0 | Ag standard |
| ii | Ag | BH4− | 92.8 | CVG loss assessment |
| iii | Ag | CO32− | 100.5 | Spray loss assessment |
The mean values for 5 or 6 parallel samples using each treatment were obtained and the relative standard deviation was 1.2–3.4%.
Results and discussion
Batch Ag chemical vapour generation
The miniature batch system, analogous to that of the ref. 4, was used for initial testing of the Ag chemical vapour generation and atomization performance, in connection with both the MMQTA atomizer and a miniature diffusion flame. The sensitivity of the batch MMQTA system (expressed as peak area characteristic mass, m0) was assessed from the peak half-width and height to be 0.6 ng, which is in good agreement with ref. 4, which reported an m0 of 0.49 ± 0.02 ng for an externally heated quartz tube atomizer of similar dimensions as our MMQTA. However, the signals were noisy and not very reproducible, probably due to the formation of bubbles during the reaction. This effect appeared also when measuring selenium, which was employed as a “reference” analyte. The only difference was that selenium produced sharper peaks; the peak half-width was around 1.8 s compared to about 3.7 s for silver.The batch mode generator was also tested with the miniature diffusion flame in order to simplify the system. The miniature diffusion flame is known to produce a high concentration of H radicals within the whole observation volume, resulting in excellent resistance towards atomization interferences.10 Thus, good atomization efficiency can be expected. On the other hand, the sensitivity is reduced due to the short optical path. The sensitivity observed (m0 = 12 ng Ag) was 20 times lower than in the MMQTA, which corresponds roughly to the selenium sensitivity ratio in both atomizers.
Silver release efficiency tested by INAA
The efficiency of the Ag release in the batch system was tested by INAA. The results of the determination of residual Ag in solution are summarized in Table 1. The results are related to a reference, row i, containing the original amount of silver in the sample and also the same amount of boron as the other treatments. Boron is known to absorb activating neutrons efficiently, which may lead to lower 110mAg activities (the Ag standard without boron exhibited a 2.2% higher activity). Row ii of Table 1 shows the results for the Ag reaction with NaBH4, i.e., the actual CVG procedure. Row iii shows the treatment that is imitating the actual physical behaviour of the reaction; NaBH4 was replaced by the amount of NaHCO3 that produces the same volume of gas by reaction with acid, but without the reducing properties.Table 1 shows that there is a removal of 7% of the analyte due to chemical vapour generation by tetrahydroborate(III) reduction. There is no other possible explanation for the losses. Spray formation can be excluded, as no loss was observed when using the treatment that produced the gas (row iii of Table 1). This confirms the findings of other authors, that spray transfer to an atomization environment is not responsible for the observed signal.4,5,7,8 The Ag release efficiency of 7% disagrees considerably with the Ag release efficiency of 80–95%, estimated indirectly from measurements of residual analyte concentrations in solution after the CVG reaction.4 However, the Ag release efficiency determined by INAA corresponds to the 8% overall efficiency estimated from sensitivities of flame AAS, as reported in ref. 4.
Continuous flow Ag chemical vapour generation
Several approaches to the design of the continuous flow-CVG system have been tested; the most satisfactory solution is shown in Fig. 1. The efficient sample–reductant mixing, the rather long reaction times and immediate volatile compound release from solution were the basic premises of its development. The fast mixing is obtained by using three concentric capillaries for the introduction of the sample, reductant and purge gas. The reaction mixture immediately enters the GLS allowing fast separation of the volatilized analyte from the liquid. However, the reaction mixture flows over the sloping wall of the GLS, which allows a longer time to complete the reaction.The most problematic points concerning the Ag CVG system are the signal stability and reproducibility. Therefore, the conditions leading to the most stable results were chosen as an optimum rather than maximum sensitivity (see below). Optimized signals are reproduced in Fig. 2(a) and (b), respectively, for diffusion flame and MMQTA.
In an earlier version of the CF system (not shown, but basically identical to that in Fig. 1), all three capillaries could be moved so that the position of the capillary end inside the GLS (D0—see Fig. 1), the distance between the gas introduction point and the end of the outer capillary (D1) and the distance between the reductant–sample mixing point and carrier gas introduction point (D2) could be independently altered. The optimum length of the outermost capillary protrusion into the GLS (D0) appeared to be 1 mm, so that the reaction mixture could flow on the wall without dropping; at D0 > 5 mm (i.e., the capillary is 5 mm inside the GLS), the capillary tip came too close to the GLS gas outlet and too many droplets entered the diffusion flame/transfer line. Changes in the individual capillary tip distances ( D1 and D2—see Fig 1) of up to 20 mm influenced the sensitivity by up to about 20%, but no systematic trends could be traced, as the original sensitivity was often not established after reversing the change. It appears that the minute differences in mutual geometric position of the capillaries at the mixing point play a role rather than the distance of the tips (or the volume of the reaction chamber). Therefore, a more rigid construction of the system was employed (Fig. 1), by fixing the capillary tips at D1 = 13 mm and D2 = 5 mm.
The level of the residual liquid in the GLS also affected the signal. The most reproducible signals were obtained when the waste outlet capillary was close to the bottom of the GLS (no liquid remaining inside the GLS). If a 3 mm level of residual liquid was used, the sensitivity was increased by about 20%, but the signal fluctuations were doubled. In the case of a maximum liquid level of 5 mm, the sensitivity was about 4 times higher. However, the signal reproducibility was very poor and pronounced wash-out times (about 5 min.) were needed. This observation suggests that volatile compound release from solution is not a fast process, which is of particular importance for further development of the CVG system.
Preliminary tests concerning the influence of individual CVG parameters were performed. The most significant observation was that the signal intensity, although directly proportional to the analyte concentration in the sample, was independent of the pumping rate of solutions (and therefore of the increased analyte supply) in the range of 0.4 to 1 ml min−1; 0.6 ml min−1 was chosen as optimum. This suggests that the formation of the volatile Ag compound is not complete under the employed conditions and/or that the limiting step is the release from solution. Changing only the flow rate of the reductant solution had no influence, except for a slight (up to 10%) signal increase at lower flow rates, probably due to lower amount of hydrogen produced. No significant change was observed if the reductant solution was prepared in 0.1–0.4% (m/v) KOH solution. For a 0.01% KOH solution, 15% lower sensitivity and noisy signals were observed. Preparing the reductant solution in 0.5 M KI resulted in a signal decrease to about 10%.
The apparent influence of the surface of the GLS was observed. At the beginning of the measurements, it took 20–30 min to establish a stable sensitivity. Visually, the glass surface was wetted without individual droplets after this period.
After prolonged use of the system with Ag concentrations at the mg l−1 level, black deposits could be observed on the walls of the GLS (mostly in the upper part where the reaction mixture enters), on the waste tubing, and especially on the tips of the capillaries where the actual reaction takes place. The deposits did not disappear by simple generation or pumping of the blank solution (1 M HNO3).
Short temporary (peak-shaped) increases in the signal (at a height of about 50% of the actual Ag signal) were occasionally experienced. These peaks were found to be caused by the bubbles in the tetrahydroborate flow. There was no difference if the bubbles contained hydrogen (from NaBH4 decomposition), or air (after a brief removal of the NaBH4 inlet capillary from the stock solution). However, an attempt to increase the signal by a constant flow of bubbles was not successful. The exact source of the observed peaks remains unclear. Most probably some enrichment of the Ag occurs close to the mixing point during the short interval of NaBH4 intake and, perhaps, the longer build-up of the Ag-containing deposits may play a role. To avoid these peaks, we successfully employed a small Teflon degassing chamber (see Fig. 1; inner volume approximately 0.5 ml) on the reductant line between the pump and the CVG system. The flow of reductant solution entered this chamber from the upper end and exited at the bottom; the bubbles entering the chamber were retained in its upper part. The chamber was refilled by inverting it for a short time.
The obtained peak area characteristic masses for the CF system were similar to those for the miniature batch system, i.e., 7 and 11 ng for sample flow rates of 0.6 and 1 ml min−1, respectively, for the diffusion flame atomizer and 0.6 ng (sample flow rate, 1 ml min−1) for the MMQTA.
From a comparison of characteristic masses for the individual arrangements, two important conclusions concerning the efficiencies of the individual steps can be drawn. Firstly, the CVG efficiency of the CF-MMQTA system is similar to that of the batch MMQTA system, since the same characteristic masses (0.6 ng) are obtained. The transport efficiency in the batch CVG-MMQTA and CF-CVG-MMQTA arrangements should be the same since the connection line is the same in both cases. The release efficiency in the batch system should be close to the 7% release efficiency observed by using INAA (see previous section), because of the similarity of the experimental set-up employed for INAA and for the batch system. Consequently, the release efficiency of the CF system should be close to these at 7%. The CF release efficiency is further increased with a decreasing sample flow rate (i.e., to around 11% at 0.6 ml min−1 sample flow rate), as lower characteristic masses are obtained (see above).
Secondly, transport losses are probably not very pronounced. In the CF-CVG-diffusion flame system, there should be no transport loss since atomization proceeds close to the GLS outlet. In the case of the batch CVG-diffusion flame system the losses are below 10% of the released analyte, which follows from the comparison of the characteristic masses for the batch CVG-diffusion flame system (employing a connection line to the atomizer, see Experimental) and the CF-CVG-diffusion flame system (no connection line) of 12 and 11 ng, respectively.
This is further supported by the fact that increasing the PTFE connection line between the CF system and the MMQTA from 150 to 280 mm (id 1 mm) reduced the signal only by 5%. On the other hand, there was a detrimental influence of the droplets (originated from the spray or water vapour condensation) in the transfer line, which impaired the reproducibility of measurement. This was the primary reason for using a diffusion flame atomizer, which can be attached without any transfer line and gives a dramatic improvement in signal stability when the originally used 80 mm PTFE transfer line (1 mm id) to the diffusion flame is rejected.
The relative limits of detection were assessed to be 0.015 µg ml−1 and 0.33 µg ml−1 for CF-CVG- MMQTA-AAS and CF-CVG-diffusion flame-AAS systems. However, it should be highlighted that the optimization was carried out primarily for the CVG step. AAS was used just as a convenient tool to develop the CVG system. No special attention was therefore paid to the minimization of the AAS limits of detection.
Conclusions
The described continuous flow chemical vapour generation of silver works well in combination with a miniature diffusion flame atomizer, yielding low noise and reproducible signals. This opens the possibility of using sensitive atomic fluorescence spectrometry for detection. The estimated 7% silver CVG efficiency compares well with the efficiency of conventional nebulizers, which confirms the potential of CVG of Ag by tetrahydroborate reduction for use with other methods of analytical atomic spectroscopy.Unfortunately, the efficiency of CVG of Ag is low compared with the classical hydride-forming elements, which is reflected in the rather poor long-time signal stability. Further CVG efficiency improvement could be achieved by careful optimization of the system design and generation parameters and/or by chemical modification.
The improvement of the volatile compound release appears to be a crucial step. However, this task is difficult without a deeper insight into the nature of the generated volatile compounds, as the identity of the volatile species remains unclear. Hypothetical hydrides, cautiously mentioned in previous reports,4,6 are one possibility. Another may be metallic “nanoparticles”, known from the work in the field of material science, produced by the tetrahydoborate reduction of solutions for a number of elements.11 The identification of the volatile species together with further optimization of the CVG system for other transient and noble metals are the obvious aims of further research.
Acknowledgements
This work was supported by a grant from GACR, No. 203/01/0453. The authors are indebted to Ralph Sturgeon for fruitful discussions.References
- J. Dědina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, Wiley, Chichester, UK, 1995 Search PubMed.
- H. G. Infante, M. L. Fernandez Sanchez and A. Sanz-Medel, J. Anal. At. Spectrom., 1999, 14, 1343 RSC.
- C. Vargas-Razo and J. F. Tyson, Fresenius' J. Anal. Chem., 2000, 366, 182 CrossRef CAS.
- A. S. Luna, R. E. Sturgeon and R. C. de Campos, Anal. Chem., 2000, 72, 3523 CrossRef CAS.
- P. Pohl and W. Zyrnicki, Anal. Chim. Acta, 2001, 429, 135 CrossRef CAS.
- R. E. Sturgeon, J. Liu, V. J. Boyko and V. T. Luong, Anal. Chem., 1996, 68, 1883 CrossRef CAS.
- C. Moor, R. E. Sturgeon and J. W. Lam, J. Anal. At. Spectrom., 2000, 15, 143 RSC.
- X. Guo, B. Huang, Z. Sun, R. Ke, Q. Wang and Z. Gong, Spectrochim. Acta, Part B, 2000, 55, 941 CrossRef.
- T. Matoušek, J. Dědina and A. Selecká, Spectrochim. Acta, Part B, 2002, 57, in the press Search PubMed.
- A. D' Ulivo and J. Dedina, Spectrochim. Acta, Part B, 1996, 51, 481 CrossRef.
- N. Panichev and R. E. Sturgeon, Anal. Chem., 1998, 70, 1670 CrossRef CAS , and references cited therein.
| This journal is © The Royal Society of Chemistry 2002 |