Solid-state synthesis of heterocyclic hydrazones using microwaves under catalyst-free conditions†
Marjan Je![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) elnika, Rajender S. Varma*b, Slovenko Polanca and Marijan Ko
elnika, Rajender S. Varma*b, Slovenko Polanca and Marijan Ko![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) evar*a
evar*a
aFaculty of Chemistry and Chemical Technology, University of Ljubljana, Akereva 5, SI-1000, Ljubljana, Slovenia. E-mail: marijan.kocevar@uni-lj.si
bClean Processes Branch, National Risk Management Research Laboratory, U.S. Environmental Protection Agency, MS 443, 26 W. Martin Luther King Drive, Cincinnati, OH 45268, USA. E-mail: varma.rajender@epa.gov
First published on 29th January 2002
Abstract
The reactions of neat 5- or 8-oxobenzopyran-2-ones, 1–3, with a variety of aromatic and heteroaromatic hydrazines, 4, are accelerated upon irradiation in a household microwave oven in the absence of any catalyst, solid support or solvent. The approach provides an attractive and environmentally friendly pathway to several synthetically useful heterocyclic hydrazones.
Green ContextAlternative energy sources and solvent free synthesis are key technologies in the green chemistry toolkit. In this article these are combined in the solid state synthesis of heterocyclic hydrazones in a household microwave oven. The reactions are fast and high-yielding.JHC |
Introduction
Microwave (MW) irradiation has been used for the rapid synthesis of a variety of compounds.1 Chemical reactions are accelerated because of selective absorption of microwave energy by polar molecules, non-polar molecules being inert to the microwave dielectric loss. Among them, heterogeneous reactions facilitated by supported reagents on various mineral oxides have received special attention in recent years.2 Relatively little attention is, however, paid to solventless reactions with neat reactants in the absence of a catalyst or solid support.3Hydrazones are important synthons for several transformations4a–d and their syntheses from various precursors are well documented.4a,b Recently, hydrazones have been prepared from carbonyl compounds and hydrazine hydrate in ethylene glycol5a and toluene5b by the application of microwave irradiation while some others are synthesized in the presence of silica gel and sodium hydroxide.6
The synthetic efforts to this class of compounds have been previously directed for the selective design of the benzopyran-2-one ring at the positions 5 and 8 as well as for the new transformation into the corresponding quinoline derivatives.7a–f The conformational analysis has been accomplished for some of these derivatives via molecular modelling techniques based on experimentally determined NOE distance restraints in order to establish their structure in solution.7a,d These studies have been stimulated, in part, due to potentially significant biological activity associated with a variety of 2H-1-benzopyrans and quinolines.7d In general, heterocyclic systems encompassing pyran unit have found application as pharmaceuticals, agrochemicals, veterinary products, dyes or pigments.7g 5,6,7,8-Tetrahydro-2H-1-benzopyran-2-ones display a wide range of biological activities, such as anti-inflammatory, local anesthetic, platelet antiaggregating, analgesic, etc.7h,i Consequently, we believe an expeditious and selective transformation of such compounds under environmentally benign conditions is of importance.
Results and discussion
For the aforementioned reasons and in view of our general interest in the development of environmentally friendlier synthetic alternatives using microwaves,2 we became interested in an expeditious synthesis of these compounds. We report here a general synthesis of several hydrazones 5–16, whose earlier preparations required relatively strenuous reaction conditions namely heating substrates for several hours in the presence of an acidic catalyst.7a–f In all the reactions reported in this paper, we worked under solvent-free conditions utilizing neat starting materials, 2H-1-benzopyran-2-ones 1,8a,b2,8a,b38c and hydrazines, 4, wherein the reactions are completed within minutes and in high yields (61–98%, Scheme 1, Table 1) using an unmodified household microwave oven. Interestingly, reactions proceeded well even when both the starting reactants were solids and the reaction temperature was maintained below the melting points of both components. Recently, some comments have been made on the preparation of phthalimides from solid reactants, phthalic anhydride and amino compounds9a with a specific MW effect being highlighted in the synthesis of fused pyrazoles.9b After the appearance of our report,3b we became aware of another investigation on conventional solid/solid organic reactions.9c In view of the unprecedented nature of this reaction, we examined the synthesis of the products 9, 11 and 15 in detail by varying the amount of hydrazines and exposure time to microwaves. In contrast to commercial microwave devices available that provide adequate mixing and control of reaction temperature, an unmodified household MW oven is used in this study with no accurate temperature control. Consequently, we decided to measure the temperature of the alumina bath that housed the reaction vessel in the MW oven with a calibrated thermometer immediately after completion of the reaction. In a controlled experiment, concurrently, we also heated the lower melting reactant in a separate glass beaker to obtain visual information about the integrity of the solid materials upon exposure to microwaves.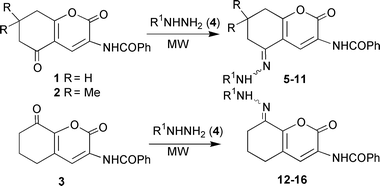 | ||
| Scheme 1 | ||
| Starting benzopyran | Hydrazine 4 (R1, mmol) | React. time/min | Product (yield, %)a |
|---|---|---|---|
| a Yield of pure product.b Reagent contains 10–15% of water for stabilization. | |||
| 1 | Ph (1.2) | 6 | 5 (83) |
| 1 | 2,5-F2C6H3 (3.0) | 8 | 6 (69) |
| 1 | 3-CF3C6H4 (1.5) | 8 | 7 (75) |
| 1 | 6-Chloropyridazin-3-yl (2.0–3.0) | 5.5 | 8 (62) |
| 2 | Ph (2.0) | 8 | 9 (61) |
| 2 | 3-CF3C6H4 (3.0) | 10 | 10 (62) |
| 2 | 6-Chloropyridazin-3-yl (2.0) | 6 | 11 (85) |
| 3 | Ph (3.0) | 3 | 12 (95) |
| 3 | 3-CF3C6H4 (1.5) | 8 | 13 (92) |
| 3 | Pyridin-2-yl (3.0) | 5 | 14 (65) |
| 3 | 4-NO2C6H4 (∼2.6)b | 9 | 15 (98) |
| 3 | 2,5-F2C6H3 (3.0) | 8 | 16 (96) |
For the synthesis of 11, the starting benzopyran 28a,b (mp 179–179.5 °C) and 3-chloro-6-hydrazinopyridazine10a (mp 135–137 °C), were admixed in the ratio 1∶2 (Table 1). When the reaction mixture was irradiated with microwaves for 5.5 min, the highest temperature in the alumina bath recorded was 120–122 °C. The reaction mixture remained solid and no reaction was observed. After heating for 6 min, however, the temperature of the bath reached 130–132 °C and the reaction mixture was completely in liquefied form affording hydrazone 11 in 85% isolated yield (see Table 1). The 3-chloro-6-hydrazinopyridazine kept in the same bath barely melted (∼5% as estimated visually) at that temperature. With the critical temperature information in hand, we embarked on a comparative study and heated the above reaction mixture in the same proportions using a preheated oil-bath. Heating for 12 min at 133 °C gave a mixture (liquid with some suspended solid material). The NMR analysis revealed that the molar ratio between hydrazone 11 and starting benzopyran 2 was approximately 3.3∶1. Prolonged heating of the reaction mixture for 1 h at 133 °C resulted in the formation of the liquid with no suspended solids. The work-up (as in general procedure) and NMR analysis revealed that the ratio between 11 and 2 in the isolated product was 92∶8. Obviously, the unreacted benzopyran derivative was still present in high enough quantity to be separated together with the product 11, while in the corresponding MW experiment, we observed complete conversion of the reactants to a single product (Table 1).
For the synthesis of hydrazone 8, the effect of the increased amount of hydrazine was evaluated. The reactants, benzopyran 18a,b (mp 188–189 °C) and 3-chloro-6-hydrazinopyridazine10a (mp 135–137 °C), were admixed in the ratio 0.5∶1.5 mmol and were subjected to microwave exposure for the same period (5.5 min) in the similar temperature range of 125–126 °C. The reaction mixture became a spongy brown mass with molten spots showing that melting had taken place followed by the formation and the separation of the solid product 8. The 3-chloro-6-hydrazinopyridazine kept in the same alumina bath barely melted (2–3% as estimated visually) at that temperature. Additional experiments with increased exposure time to microwaves (alumina bath temperature being ∼133 °C) resulted in the development of fumes and a darkened reaction mixture with hydrazone 8 being detected by TLC. In a separate experiment, we irradiated in the same bath 3-chloro-6-hydrazinopyridazine and benzopyran 1 (in different beakers) and both remained practically unchanged (pyridazine derivative slightly melted). The repetition of the above experiments with the ratio of the starting compounds, benzopyran 2∶3-chloro-6-hydrazinopyridazine being 1∶2, resulted in a spongy brown mass that afforded the product 8 in 62% isolated yield (Table 1). Even extended irradiation for up to 7 min (∼135 °C) with this ratio (1∶2) resulted in no observable decomposition of the reaction mixture. With two successive irradiations of the same reaction mixture (5 min each, bath temperature ∼150 °C), no violent decomposition was observed. These experiments indicate the importance of the correct ratio of the reactants; higher amounts of hydrazine are detrimental as demonstrated in the uncontrollable reaction when the reactants are in the ratio 1∶3. A comparative study was performed using a preheated oil-bath at 128 °C for 20 min. The NMR analysis after the work-up (as in the general procedure) revealed that the molar ratio between hydrazone 8 and starting benzopyran 1 was approximately 2.6∶1.
For the synthesis of 15 starting from 38c (mp 248–250 °C) and 4-nitrophenylhydrazine10b (mp ∼157 °C with decomposition) we performed an experiment with the reactants in the ratio 1∶2.6, respectively. The commercially obtained hydrazine derivative, however, contained 10–15% of water for stabilization and as such slightly melted at 130 °C. Upon MW irradiation for 5.5 min (120–122 °C), the reaction mixture was completely melted while the 4-nitrophenylhydrazine in the second beaker was only barely melted. NMR analysis of crude mixture showed no presence of the benzopyran derivative 3, and the work-up resulted in hydrazone 15 in 94% yield. On the other hand, heating the same reaction mixture for 1 h at 128 °C in an oil-bath resulted in a mixture of product 15 and starting 3 in the ratio 4.3∶1, as discerned from 1H NMR spectrum of the crude reaction mixture. The reaction mixture at the end of this heating period was solid. This comparative study of the reactions taking place in an oil-bath in the absence of MW heating revealed a substantial rate enhancement for reactions conducted under MW irradiation conditions, presumably due to the increase in polarity after change from the solid to the liquid phase.9a Such bimolecular reactions2d,e will show a polarity enhancement as a result of the ensuing intermediate transition state and consequently may display a pronounced microwave effect.9b Further, the reactions could be visibly monitored since no reaction occurs without formation of a melt. This methodology allows for performing rapid syntheses of a variety of hydrazones below the melting points of the participating starting materials. This could be explained by lowering of the melting point by the formation of the eutectic.11 Consequently, such methodology seems to be especially useful when starting from substrates, which decompose at the normal melting point.
An adequate temperature control of the reaction, however, is strongly recommended. The temperature of the alumina bath upon MW heating for a specific period is dependent on the moisture (water) content of alumina. As an example, dried and desiccated alumina due to the diminished water content may heat up to 20 °C lower than the alumina in a bath stored in the laboratory atmosphere. The consequence of the lower temperature may influence the outcome of the reaction; solid reactants may not melt and no reaction may be observed.
Conclusion
In conclusion, we have developed an expeditious, easy-to-handle and environmentally friendlier approach to the synthesis of a variety of non-easily-available hydrazones using microwave irradiation that can be extended to other systems.Experimental
Melting points were determined on a Kofler micro hot stage, and are uncorrected. 1H (300 MHz) and 13C NMR (75 MHz) spectra were recorded with the Bruker Avance DPX 300 and Varian VXR-300 Unity Plus spectrometers in DMSO-d6, using TMS as an internal standard. The coupling constants (J) are given in Hz. IR spectra were obtained with a Perkin Elmer Spectrum 1000 spectrophotometer. Mass spectra were recorded with a VG-Analytical AutoSpec Q instrument. Elemental analyses (C, H, N) were performed with a Perkin Elmer 2400 CHN Analyzer. Thin-layer chromatography was carried out on Fluka silica gel TLC-cards. The starting benzopyran-2-ones, 1–3, and 3-chloro-6-hydrazinopyridazine were prepared as described in the literature.8,10a All other reagents and solvents were used as received from commercial suppliers. An unmodified household microwave oven operating at 2450 MHz was used at its full power, 650 W, for all the experiments.General procedure for the synthesis of hydrazones 5–16
A neat mixture of benzopyran derivative 1–3 (1 mmol) and hydrazine 4 (1.2–3 mmol; see Table 1) in a 10 mL glass beaker was thoroughly mixed for about 5 min, then it was placed in an alumina bath inside the household microwave oven and irradiated. The maximum temperature reached in the alumina after 10 min was about 150 °C. After cooling, methanol (∼4 mL) was added to the mixture and the separated solid was filtered off and washed with a small amount of methanol. The products were crystallized from appropriate solvents for elemental analysis. The details of reaction conditions and yields are provided in the Table 1.Analytical and spectroscopic data of products 5–16
Acknowledgements
The authors wish to thank the Ministry of Education, Science and Sport of the Republic of Slovenia for financial support (P0-0503-103). Dr. B. Kralj and Dr. D.![[Z with combining breve]](https://www.rsc.org/images/entities/char_005a_0306.gif) igon (Center for Mass Spectroscopy, ‘Jo
igon (Center for Mass Spectroscopy, ‘Jo![[z with combining breve]](https://www.rsc.org/images/entities/char_007a_0306.gif) ef Stefan’ Institute, Ljubljana, Slovenia) are gratefully acknowledged for mass spectra measurements, and Dr J. Plavec for NMR spectroscopic measurements and helpful discussion concerning some NMR spectra.
ef Stefan’ Institute, Ljubljana, Slovenia) are gratefully acknowledged for mass spectra measurements, and Dr J. Plavec for NMR spectroscopic measurements and helpful discussion concerning some NMR spectra.References
- (a) S. Caddick, Tetrahedron, 1995, 51, 10403–10432 CrossRef; (b) R. S. Varma, Green Chem., 1999, 1, 43–55 RSC; (c) R. S. Varma, J. Heterocycl. Chem., 1999, 36, 1565–1571 Search PubMed; (d) A. Loupy, A. Petit, J. Hamelin, F. Texier-Boullet, P. Jacquault and D. Mathe, Synthesis, 1998, 1213–1234 CrossRef CAS; (e) A. de la Hoz, A. Díaz-Ortis, A. Moreno and F. Langa, Eur. J. Org. Chem., 2000, 3659–3673 CrossRef.
- (a) R. S. Varma, in ACS Symposium Series No. 767/ Green Chemical Syntheses and Processes, ed. P. T. Anastas, L. Heine and T. Williamson, American Chemical Society, Washington DC, 2000, ch. 23, pp. 292–313 Search PubMed; (b) R. S. Varma, in Green Chemistry: Challenging Perspectives, ed. P. Tundo and P. T. Anastas, Oxford University Press, Oxford, 2000, pp. 221–244 Search PubMed; (c) R. S. Varma, Pure Appl. Chem., 2001, 73, 193–198 Search PubMed; (d) R. S. Varma, R. Dahiya and S. Kumar, Tetrahedron Lett., 1997, 38, 2039–2042 CrossRef CAS; (e) A. Vass, J. Dudas and R. S. Varma, Tetrahedron Lett., 1999, 40, 4951–4954 CrossRef CAS; (f) R. S. Varma, Clean Products Processes, 1999, 1, 132–147 Search PubMed; (g) R. S. Varma and V. N. Namboodiri, Chem. Commun., 2001, 643–644 RSC; (h) R. S. Varma and V. N. Namboodiri, Pure Appl. Chem., 2001, 73, 1309–1313 Search PubMed.
-
(a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS;
(b) M. Jeelnik, R. S. Varma, S. Polanc and M. Koevar, Chem. Commun., 2001, 1716–1717 RSC.
- (a) J. S. Clark, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Pergamon, Oxford, 1995, vol. 3, pp. 443–490 Search PubMed; (b) D. E. Bergbreiter and M. Momongan, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 2, pp. 503–526 Search PubMed; (c) W. Sucrow, Org. Prep. Proced. Int., 1982, 14, 93–155 Search PubMed; (d) R. Fusco and F. Sannicolo, Tetrahedron, 1980, 36, 161–170 CrossRef CAS.
- (a) B. K. Banik, K. J. Barakat, W. R. Wagle, M. S. Manhas and A. K. Bose, J. Org. Chem., 1999, 64, 5746–5753 CrossRef CAS; (b) S. Gadhwal, M. Baruah and J. S. Sandhu, Synlett, 1999, 1573–1574 CAS.
- A. R. Hajipour, I. Mohammadpoor-Baltork and M. Bigdeli, J. Chem. Res., (S), 1999, 570–571 RSC.
-
(a) P. Trebe, S. Polanc, M. Koevar, T.
![[S with combining breve]](https://www.rsc.org/images/entities/char_0053_0306.gif) olmajer and S. Goli Grdadolnik, Tetrahedron, 1997, 53, 1383–1390 CrossRef CAS;
(b) P. Trebe, B. Recelj, M. Koevar and S. Polanc, J. Heterocycl. Chem., 1997, 34, 1247–1250 Search PubMed;
(c) P. Trebe, B. Recelj, T. Lukanc, S. Golic Grdadolnik, A. Petri, B. Verek, T. olmajer, S. Polanc and M. Koevar, Synth. Commun., 1997, 27, 2637–2644 CAS;
(d) S. Goli Grdadolnik, P. Trebe, M. Koevar and T. olmajer, J. Chem. Inf. Comput. Sci., 1997, 37, 489–494 CrossRef;
(e) M. Koevar, Chem. Listy, 1997, 91, 610–615 Search PubMed;
(f) P. Trebe, L. Vraniar, I. Muic, S. Polanc, W. C. Stevens and M. Koevar, Heterocycles, 2000, 53, 1111–1120 Search PubMed;
(g) G. R. Geen, J. M. Evans and A. K. Vong, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, 1996, vol. 5, pp. 469–500 Search PubMed;
(h) M. Longobardi, A. Bargagna, E. Mariani, P. Schenone, M. D′Amico, A. Filippelli, C. Falzarano and E. Lampa, Farmaco, 1993, 48, 1121–1130 CAS;
(i) L. Mosti, G. Menozzi, P. Schenone, M. D′Amico, M. Falciani and F. Rossi, Farmaco, 1994, 49, 45–50 CAS.
olmajer and S. Goli Grdadolnik, Tetrahedron, 1997, 53, 1383–1390 CrossRef CAS;
(b) P. Trebe, B. Recelj, M. Koevar and S. Polanc, J. Heterocycl. Chem., 1997, 34, 1247–1250 Search PubMed;
(c) P. Trebe, B. Recelj, T. Lukanc, S. Golic Grdadolnik, A. Petri, B. Verek, T. olmajer, S. Polanc and M. Koevar, Synth. Commun., 1997, 27, 2637–2644 CAS;
(d) S. Goli Grdadolnik, P. Trebe, M. Koevar and T. olmajer, J. Chem. Inf. Comput. Sci., 1997, 37, 489–494 CrossRef;
(e) M. Koevar, Chem. Listy, 1997, 91, 610–615 Search PubMed;
(f) P. Trebe, L. Vraniar, I. Muic, S. Polanc, W. C. Stevens and M. Koevar, Heterocycles, 2000, 53, 1111–1120 Search PubMed;
(g) G. R. Geen, J. M. Evans and A. K. Vong, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, 1996, vol. 5, pp. 469–500 Search PubMed;
(h) M. Longobardi, A. Bargagna, E. Mariani, P. Schenone, M. D′Amico, A. Filippelli, C. Falzarano and E. Lampa, Farmaco, 1993, 48, 1121–1130 CAS;
(i) L. Mosti, G. Menozzi, P. Schenone, M. D′Amico, M. Falciani and F. Rossi, Farmaco, 1994, 49, 45–50 CAS. -
(a) M. Koevar, S. Polanc, M. Tiler and B. Verek, Synth. Commun., 1989, 19, 1713–1719 CAS;
(b) V. Kepe, M. Koevar, S. Polanc, B. Verek and M. Tiler, Tetrahedron, 1990, 46, 2081–2088 CrossRef CAS;
(c) V. Kepe, M. Koevar, A. Petri, S. Polanc and B. Verek, Heterocycles, 1992, 33, 843–849 Search PubMed.
- (a) T. Vidal, A. Petit, A. Loupy and R. N. Gedye, Tetrahedron, 2000, 56, 5473–5478 CrossRef; (b) S. Paul, M. Gupta, R. Gupta and A. Loupy, Tetrahedron Lett., 2001, 42, 3827–3829 CrossRef CAS; (c) G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701–8708 CrossRef CAS.
- (a) N. Takahayashi, J. Pharm. Soc. Jpn., 1955, 75, 778–781 Search PubMed; Chem. Abstr., 1956, 50, 4970c Search PubMed; (b) The Merck Index, 12th edn., Merck & Co., Inc., Whitehouse Station, 1996, p. 1138, (Monograph number 6721) Search PubMed.
- C. F. Most, Experimental Organic Chemistry, J. Wiley & Sons, New York, 1988, pp. 53–62 Search PubMed.
Footnote |
| † Presented, in part, at the 5th Electronic Conference on Synthetic Organic Chemistry, (ECSOC-5), 1–30 September 2001, E0014, http://www.mdpi.org/ecsoc-5.htm |
| This journal is © The Royal Society of Chemistry 2002 |