Interlaboratory comparability of copper complexation capacity determination in natural waters
Eleanor
van Veen
ab,
Sean
Comber
b and
Michael
Gardner
*b
aImperial College of Science, Technology and Medicine, Silwood Park, Buckhurst Road, Ascot, Berkshire, UK SL5 7TE
bWRc-NSF, Henley Road, Medmenham, Marlow, UK SL7 2HD. E-mail: gardner_mj@wrcplc.co.uk
First published on 5th December 2001
Abstract
The results of a test of interlaboratory comparability for the determination of copper complexation capacity and copper–ligand complex formation constant are presented. Eight water samples comprising, six natural waters, a synthetic ligand solution and a blank solution were analysed by seven laboratories using their own methods of complexation titration. Given the wide variation that might have been possible, relatively good comparability was demonstrated amongst the variety of methods for determining copper complexation capacity. The complexation capacity data largely satisfied the predefined criterion of agreement to within 50%. This provides support for the use of metal speciation criteria in the regulation of copper in the environment. Data for the determination of complex formation constants were of poorer comparability, ranging between 107 and 1012 for the same water.
Background
Despite the fact that for many years it has been accepted that speciation is a major influence in determining the environmental impact of metal pollution, the concept of metal speciation has not been applied widely in managing the water environment. One of the more important reasons for this is that non-specialists see the determination of key speciation characteristics as complicated, somewhat arbitrary and possibly contentious. This is a reflection of the fact that such determinations are, in metrological terms, relatively immature. That is, they are usually carried out in a research context, they have not been adopted as parameters for routine environmental monitoring and, most importantly, they tend not to have been shown to be adequately comparable or reliable in order to be accepted by regulatory agencies as the basis for pollution control.Copper is perhaps the most important metal for which aqueous speciation is critical to ascertain potential environmental impact. There are three main reasons for this. Firstly, copper is very widely used. Many uses can lead to its occurrence in surface waters at concentrations of potential importance to aquatic life. Secondly, copper is known to exhibit an affinity for natural complexing agents1–5 and, thirdly, organically complexed forms of the metal have been shown to be substantially less toxic than “free” or inorganic species.
The two principal factors that define copper speciation in natural water are the effective total ligand concentration, the complexation capacity (CC), and the conditional formation constant, K′, of the copper–ligand complex in the water matrix of interest.
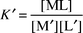 | (1) |
This paper describes an interlaboratory test for the determination of the copper CC of natural waters. Eight water samples comprising, six natural waters, a synthetic ligand solution and a blank solution were analysed by seven laboratories using their own methods of complexation titration. Our intention in undertaking this work was to provide an illustration of the comparability of determinations of copper CC, to prompt consideration of the need to harmonise such measurements and to help to evaluate the confidence that data users might place in such measurements.
Experimental design and sample preparation
Around a dozen laboratories were invited to take part in the test. These were chosen on the basis that they had published work on copper CC and were known to be likely to be interested in a test of this nature. The two key issues to be agreed at the outset were “what is the determinand?” and “what fitness for purpose criteria should be used to judge the degree of comparability that the test would reveal”. With respect to the former issue, it was clear that research techniques developed in different laboratories to assess copper complexation in general might not necessarily address exactly the same copper species. Hence it was recognised that this interlaboratory study was essentially one of comparability between laboratories/techniques, rather than a check on absolute accuracy, for a clearly defined determinand. However, in order to establish a rationale for the determination of CC, it was agreed that the overall objective was to determine complexation by organic ligands that might be considered to offer biota a significant level of protection from copper toxicity. With respect to fitness for purpose criteria, it is not clear exactly how CC data might be used in detail, though it might be envisaged that the lower toxicity of complexed forms might be used to produce a more risk-based approach to the regulation of copper contamination in natural waters. With this in mind, we decided that agreement to within a factor of two might be regarded as the basis of relatively good comparability.Table 1 summarises the nature and sources of the interlaboratory test samples. Portions (25 l) of each of the natural waters were collected and filtered to 0.2 µm using a high capacity cartridge filter (Sartorius, Watford, UK) and a peristaltic pump. The filtered samples were thoroughly mixed and dispensed into 500 ml low density polyethylene bottles (Nalgene, UK) that had been filled for 3 days with 5% v/v nitric acid (Aristar, Merck, Poole, UK) and rinsed three times with deionised water. Sample filtration marked the first day of the test. Samples were dispatched to participating laboratories on day 2. Participants were asked to carry out two replicate determinations of total copper CC on each sample. It was pointed out that total CC included the CC associated with the background copper concentration in the sample—hence that it would be necessary to determine copper concentrations. Brief details of the speciation technique were requested on the results form. Participants were told that the water samples were expected to be of CC in the range 50–500 nM (based on the organisers' preliminary analyses). They were asked to analyse the samples within 10 days and to report results, within 4 weeks, for as many samples for which they felt their technique was suitable. Background data on the nature of the samples were also provided to avoid participants having to obtain such data for themselves, so adding to the burden of taking part. These data are listed in Table 2.
| Sample | Descriptiona |
|---|---|
| a ppt = parts per thousand. b EPPS = N-2-hydroxyethylpiperazine-N'-3-propane sulfonic acid. | |
| A: River Goyt, Derbyshire | Upland river water, soft, coloured |
| B: River Test, Hampshire | Lowland river water, hard, unpolluted |
| C: River Thames Buckinghamshire | Lowland river water, hard, polluted |
| D: River Humber, Hull, Yorkshire | Estuarine water (salinity 6 ppt) |
| E: River Humber, Hull, Yorkshire | Estuarine water (salinity 12 ppt) |
| F: St. Davids, Pembrokeshire | Seawater (35 ppt) |
| G | EDTA solution (buffered to pH 8 with EPPSb) |
| H | Buffered blank (pH 8, EPPS) |
| Sample A | Sample B | Sample C | Sample D | Sample E | Sample F | |
|---|---|---|---|---|---|---|
| a Data for freshwaters are measured. For saline waters DOC and pH are measured, the remaining parameters are inferred from salinity and river water quality. nd = not determined. na = not applicable. b Dissolved organic carbon. c Electrical conductivity. | ||||||
| Calcium/mg l−1 | 2.5 | 102 | 96 | 145 | 189 | 360 |
| Sodium/mg l−1 | nd | nd | nd | 1749 | 3489 | 10157 |
| Chloride/mg l−1 | nd | nd | nd | 3143 | 6268 | 18247 |
| Magnesium/mg l−1 | 2 | 1.8 | 4.4 | 211 | 415 | 1200 |
| Potassium/mg l−1 | nd | nd | nd | 62 | 122 | 351 |
| DOCb/mg l−1 | 12.1 | 2 | 5.1 | 5.1 | 4.2 | <1 |
| pH | 4.3 | 8.1 | 8.1 | 8 | 8 | 8 |
| Elec cond.c/µS cm−1 | 142 | 595 | 573 | na | na | na |
| Salinity (ppt) | na | na | na | 6 | 12 | 35 |
It was recognised that sample stability was one of the critical factors to the validity of the test. If participants analysed portions of sample that were substantially different owing to changes in CC that had taken place after distribution, the test would not show true comparability. A preliminary evaluation of the stability of a representative water was therefore undertaken. CC was determined on six occasions over a 20 day period on a filtered lowland river sample taken from the same source as Sample C used subsequently in the interlaboratory test. The sample was stored in the dark at room temperature. The results of this preliminary test indicated no significant change in CC, where an overall change of approximately 15% would have achieved significance. On this basis, it was decided to proceed with the test, but to carry out a detailed concurrent study of the stability of all the distributed samples. This involved duplicate determinations of CC on each sample [using the cathodic stripping voltammetric (CSV) methodology employed at the organisers' laboratory] on up to five occasions, over a period of 30 days. Samples were stored in the dark at room temperature for the first 5 days (a longer time than the samples took to reach all but one participant) and then in the dark under refrigeration for the remaining time.
Results
The results of the test of CC stability are shown in Fig. 1. No significant changes were observed, apart from for the final 30 day analysis of Sample G. A significant trend was taken to be a cumulative change as large as the pooled confidence limit over three points or a change as large as twice the pooled confidence limit for one point. The power of the test to detect trends varied from one sample to another between 10 and 30 nM (apart from Sample A where the smallest detectable change was closer to 50 nM). This variation was largely an inverse function of the limiting slope of the plot of peak current versus concentration. Methodologies are summarised in Table 3. Data for reported concentrations of dissolved copper are shown in Table 4. The results of laboratories' determinations of CC are summarised in Fig. 2. Data reported for log K′ values are shown in Fig. 3. Seven laboratories took part in the test. The different methods they used for the determination of CC concentration are summarised in Table 3.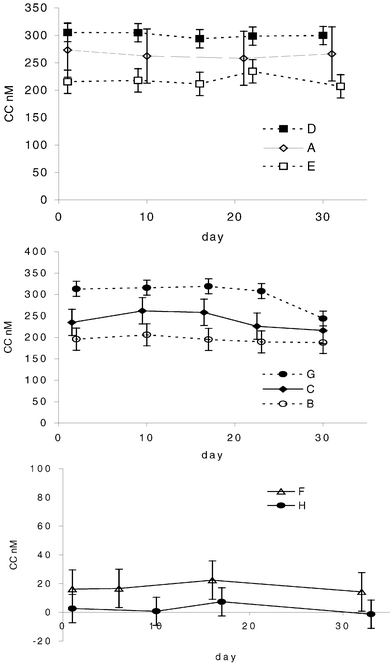 | ||
| Fig. 1 Tests of sample stability. Copper CC (nM) by CSV (catechol at 10−5 M) versus sample age in days. Samples are identified by their letter. Error bars show 95% confidence intervals of the mean of duplicate titrations on a given day based on a pooled, within-sample standard deviation with 4 or 5 degrees of freedom. Day 1 was the day of sample filtration and bottling. Samples were dispatched on day 2. Data for Sample A were obtained using anodic stripping voltammetry. | ||
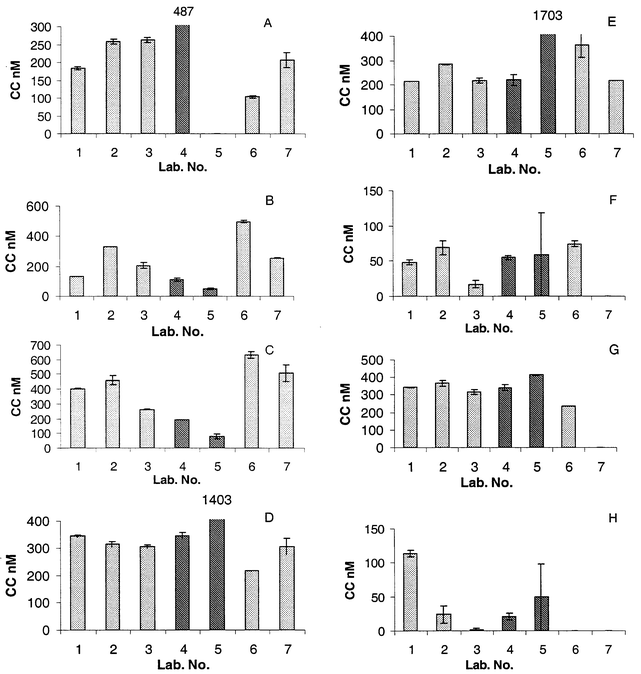 | ||
| Fig. 2 Histograms showing the comparability of data for copper CC determination. The sample identifier letter is in the top left hand corner of each histogram. Laboratories using electrochemical speciation techniques are indicated by the light shading in the columns. The two participants using techniques based on resin adsorption are indicated by darker shading. The error bars show the range of two replicate results, where duplicate data were reported . Where no results are shown, no results were reported. | ||
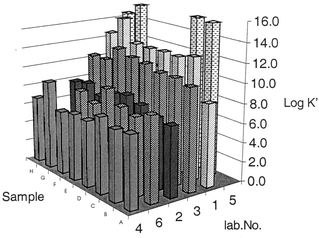 | ||
| Fig. 3 Comparability of nominal log formation constants for the copper–ligand complexes. | ||
| Lab. | Method summary |
|---|---|
| 1 | Sample A: ASV at pH 4.0 (phthalate buffer) 10 titration points each of addition 50 nM Cu. |
| Samples B–H CSV with salicylaldoxime (5 µM) at pH 8.3 (borate buffer) or EPPS (pH 8.0) for sample G and H. | |
| Samples B, E, H: 10 points each of addition 50 nM Cu. | |
| Samples C, D, G: 10 points each of 100 nM Cu. | |
| Sample F: 10 points each of 20 nM Cu. | |
| 2 | ASV with samples B–H buffered with EPPS at pH 8.0 (Sample A unbuffered). Titrations involve 10 points—the first six additions of 80 nM, the remainder of 160 nM. 4 min equilibration after each addition to a single 15 ml portion of sample. |
| 3 | Sample A: ASV at pH 4.0 (unbuffered) with 10 titration points each of addition 160 nM Cu. |
| Samples B–H CSV with catechol (1 µM) at pH 8.0 (EPPS buffer). Titrations involve 10 points—the first six additions of 80 nM, the remainder of 160 nM. 4 min equilibration after each addition to a single 15 ml portion of sample. | |
| 4 | Labile metal is sorbed to the resin Chelex 100 (100–200 mesh) in a column process involving 15 ml sub-portions of sample. Columns are approx. 40 mm long and of 2 mm diameter. Sample contact time is approx. 2 min. The complexed metal that is non-labile to Chelex sorption is determined by GFAAS. |
| 5 | The concentration of copper sorbed to the complexing resin Amberlite CG 50 is determined. A batch process is used with an equilibration time of 16 h. Eight sub-portions of the sample are used in the titration in a batch process. The sorbed labile metal is determined by GFAAS. |
| 6 | ASV with samples B–H buffered to natural (measured pH) using a nitrogen–CO2 mixture. Sample A was not buffered. Titrations involve 10 points—the first six additions of 80 nM, the remainder of 160 nM. 10 min equilibration after each addition to a single portion of sample. |
| 7 | ASV with samples B–F buffered to natural pH with borate buffer. |
| Sample | Lab. 1 | Lab. 2 | Lab. 3 | Lab. 4 | Lab. 5 | Lab. 6 | Lab. 7 |
|---|---|---|---|---|---|---|---|
| A | 40 | 36 | 44 | 39 | 39 | 38 | 85 |
| B | 16 | 31 | 16 | 19 | 12 | 11 | 62 |
| C | 28 | 58 | 54 | 47 | 36 | 27 | 73 |
| D | 77 | 77 | 79 | 71 | 129 | 76 | 187 |
| E | 55 | 68 | 71 | 66 | 112 | 45 | 142 |
| F | 4.3 | 7.9 | 6.3 | 6.3 | 33 | 8.0 | 40 |
| G | 3.8 | 16 | 19 | 6.3 | <8 | 12 | 0 |
| H | 4.1 | 7.9 | 9.4 | 6.3 | <8 | 8.0 | 17 |
Discussion
There are two principal causes of poor comparability of measurement: poor precision and bias. At high levels of CC poor precision of measurement would be evident as large differences between replicate determinations of the same sample; at low levels of CC, poor precision would lead to inability to detect CC. The duplicate data reported for this exercise did not indicate that poor precision was the main source of differences between laboratories' data.Interlaboratory bias in observed CC might arise from four main sources: (a) bias in the determination of dissolved copper concentrations; (b) bias in the concentration of the spiking standard solution used in the titration; (c) bias caused by the matrix of the sample (this type of bias should be calibrated out in the CC titration but may not be if the ligand were not titrated to full saturation or if factors such as ionic strength affect the performance of the technique); and (d) bias caused by different classes of ligands being detected to different extents by each technique. Bias in the method of data processing might be also be added to this list, though this source of bias might more usefully be included with other aspects of methodology.
The data reported for dissolved copper determinations do not indicate that bias in the determination of dissolved copper is important, though some bias is evident for Labs. 5 and 7, particularly for the saline samples. The differences are if anything smaller than have been reported in other studies of the comparability of trace metal determination.11 The good control over calibration of copper determinations evident in the good comparability might be taken to imply that error in the spiking solutions used is also small. The issue of matrix effects seems to be more significant with the two laboratories using techniques based on solid phase chelation showing larger differences for the samples of more unusual matrices, whilst achieving relatively good comparability for the other samples. In the case of Lab. 4, the largest difference is for the low pH sample; for Lab. 5. the largest deviations from the electrochemical consensus are for the two estuarine samples.
However, the main source of interlaboratory bias appears to be that the ligands accessed by different techniques are simply different [source (d), above]. Even for laboratories using the same equipment and ostensibly the same technique (e.g., Labs. 2 and 6) there are instances where notable differences occur. This type of bias would tend not to be constant from one sample to another. In a natural water it is likely that there will be a range of different copper complexing ligands. This is self-evident considering the potentially great variety of source of copper complexing substances, each of which has been shown to be important, e.g., riverine and marine biological and microbiological exudates, including proteins and polysaccharides, similar substances derived from sewage treatment, the decomposition products of sewage, natural (humic) substances, industrially produced ligands. Each of these ligand types will have a different affinity for copper and will be present at different concentrations in different water bodies.
In order to detect complexation, it is necessary to apply either a detection technique or a separation technique, either of which can differentiate between complexed and “free” metal. Differences between CC reported by different laboratories may be due to the fact that the different detection/separation techniques compete with natural complexation to different extents. For example, the technique of cathodic stripping voltammetry involves the addition of a copper complexing ligand that can adsorb to the mercury drop, to be measured as an indication of “free copper” concentration. The successful application of the technique involves the choice of a concentration for the added competitor ligand that is low enough to minimise competition with (and decomposition of) the natural complexes, but high enough to provide a measurable signal for labile metal. This involves a compromise between the achievement of adequate analytical performance and the detection of the complexes between copper and the natural ligands of weaker affinity of copper.
One fact came to light in relation to the methodology of complexation titration. There is some debate about the time allowed for equilibration between the added titrant metal and natural ligands.12 Some workers take the view that only the ligands that react to form complexes in a few minutes should be counted as exerting a protective effect to aquatic life. Hence they titrate by adding successive amounts of metal to the same portion of sample with an equilibration period of approximately 5–10 min. Others take the view that this approach will fail to detect ligands that take longer to form complexes and allow an overnight equilibration period, using a series of separate portions of sample. One participant in this study carried out titrations in both ways and reported that the two methods gave the same value to within approximately 20% for CC. This confirms a similar comparison reported earlier.13
The buffered blank gave a positive CC for several techniques. Residual CC from zwitterionic ‘biological’ buffers has been reported elsewhere.14 This emphasises the necessity of doing blanks as part of the process of CC determination (as in other types of analysis). The EDTA standard proved to be a useful, though possibly not ideal, control sample for validation of CC determination. A better control ligand would need to posses an affinity for copper that is both known and similar to that of natural ligands. As with the blank sample, data for EDTA point to the advisability of applying quality control measures to CC determination. Two participants reported that, for anodic stripping voltammetric (ASV)-based methods, deposition potential is critical to the result obtained for EDTA. It seems that at more negative values of deposition potential (>approx. −0.4V) the copper–EDTA complex may be labile.
Values reported for complex formation constants varied greatly, given the log scale that is commonly employed. It was necessary to contact participants to check that they were reporting log K′ values for the equilibrium with Cu2+ (taking account of inorganic side reactions) rather than with labile copper. Two participants needed to recalculate their log K′ values to ensure that the relevant equilibrium was:
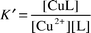 | (2) |
Conclusions
An evaluation of the comparability of the determination of metal speciation is an important part of the development of water quality criteria that truly reflect the risk posed by metals in the environment. Given the wide variation that might have been possible, relatively good comparability has been demonstrated between a variety of methods of copper CC determination. The CC data in this test largely satisfied the criterion of agreeing to within a factor of two. This provides support for the use of CC determination in the regulation of copper in the environment.Methodology for the establishment of log formation constants for copper–ligand complexes is subject to serious interlaboratory bias. This does not preclude the inclusion of aspects of copper speciation in pollution control applications, provided it can be established that the CC detected relates to complexation that is sufficiently strong to afford protection to target organisms. However, data reported for log K′ values appear to range from around 7 to 12, even though the corresponding CC data are reasonably comparable.
Acknowledgements
The participants in the test are listed below in random order. We would like to thank our collaborators, both for their considerable efforts in undertaking the test and for the substantial amount of sound advice and encouragement that we received.University of Pavia, CSIRO Sydney, Imperial College London, LAQUIPAI Porto, WRc-NSF (2), IST Lisbon.
References
- D. Tang, K. W. Warnken and P. H. Santschi, Limnol. Oceanogr., 2001, 46, 321 Search PubMed.
- S. E. J. Buykx, R. F. M. J. Cleven, A. A. Hoegee-Wehmann and M. A. G. T. van den Hoop, Fresenius' J. Anal. Chem., 1999, 363, 599 CrossRef CAS.
- M. J. Gardner, E. M. Dixon and S. Comber, Chem. Speciation Bioavailability, 2000, 12, 1 Search PubMed.
- C. M. G. van den Berg, Mar. Chem., 1984, 15, 1 CrossRef CAS.
- C. M. G. van den Berg, Mar. Chem., 1982, 11, 307 CrossRef CAS.
- R. C. Playle, D. G. Dixon and K. Burnison, Can. J. Fish. Aquat. Sci., 1993, 50, 2667 CAS.
- R. J. Erickson, D. A. Benoit, V. R. Mattson, H. P. Nelson and E. N. Leonard, Environ. Toxicol. Chem., 1996, 15, 181 CAS.
- H. E. Allen and D. J. Hansen, Water Environ. Res., 1996, 68, 42 Search PubMed.
- J. Gardiner and G. Mance, United Kingdom water quality standards arising from European Community Directives, Water Research Report, TR 204, Water Research Centre, Marlow, 1984 Search PubMed.
- E. M. Dixon, M. J. Gardner and S. J. Parry, Chem. Speciation Bioavailability, 1999, 11, 51 Search PubMed.
- Analytical Quality Control (Harmonised Monitoring) Committee, Analyst, 1985, 110, 1 RSC.
- H. Z. Ma, S. D. Kim, D. K. Cha and H. E. Allen, Environ. Toxicol. Chem., 1999, 18, 828 CAS.
- M. J. Gardner and J. E. Ravenscroft, Chemosphere, 1991, 23, 695 CrossRef CAS.
- H. Soares and P. Conde, Anal. Chim. Acta, 2000, 421, 103 CrossRef CAS.
- I. Ruzic, Anal. Chim. Acta, 1982, 140, 99 CrossRef CAS.
- S. C. Apte, M. J. Gardner, J. E. Ravenscroft and J. A. Turrell, Anal. Chim. Acta, 1990, 235, 287 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |