Normalization procedures for sediment contaminants in spatial and temporal trend monitoring
Michael Kerstena and Foppe Smedesb
aGeoscience Institute, Gutenberg-University, D-55099 Mainz, Germany
bNational Institute for Coastal and Marine Management/RIKZ, PO Box 207, NL-9750 AE Haren, The Netherlands
First published on 10th January 2002
Abstract
Rational pollution, or the effectiveness of natural attenuation assessments based upon estimating the degree of contamination, critically depends on the basis of a sound normalization to take into account heterogeneous sedimentary environments. By normalizing the measured contaminant concentration patterns for the sediment characteristics, the inherent variability can be reduced and so allow a more meaningful assessment of both the spatial distributions and the temporal trends. A brief overview and guidance in the methodology available for choosing an appropriate site-specific normalization approach is presented. This is followed by general recommendations with respect to the choice of normalizer and the necessary geochemical and statistical quality assurance methods, with support from the results of recent international intercomparison exercises within the QUASH (Quality Assurance of Sample Handling) programme, as well as discussions within the International Commission on the Exploration of the Sea (ICES) working groups. The most important of these recommendations is the use of a two-tiered normalization approach including wet sieving (<63 µm), followed by an additional geochemical co-factor normalization.
1. Introduction
The most important factor in defining the fate of and transport processes influencing contaminants in sediment is the energy of the overlying flow. In high energy environments, the sediment tends to be coarse grained and non-cohesive with little sorptive capacity and low depositional rates. In low energy environments, a significant deposition of fine-grained sediments exists, providing a high sorptive capacity for contaminants. This causes contaminant concentrations to be generally higher in areas with sediments rich in clays than those in sandy regions, where the granulometric dilution effect causes lower and variable contaminant concentrations in the sediment. Small-scale spatial variance in the concentration of pollutants in sediments may therefore be related to variance in the ambient energy of the aquatic system being investigated, which may exceed one order of magnitude.1At low-energy sites, where natural sediment burial is reasonably rapid, intrinsic natural remediation may be an acceptable alternative to excavation and off-site remediation. Continuous monitoring is necessary, however, to ensure that the conceptual and quantitative models of the efficacy of natural attenuation of contaminants are validated by spatial and temporal trend analysis and confirmed for predictive use. Problems may arise in areas where results of trend monitoring are obscured by selective transport and sedimentation effects.2 Pollution assessment and, in particular, the success of remediation activities or natural attenuation are also challenged by a need to evaluate contaminant levels using site-specific background levels from sediments of different origins with different textural, physicochemical, and compositional characteristics. Without correction for variable contaminant background levels and uptake capacity, meaningful comparisons of contaminant concentrations are impeded by large levels of bias and variability, even in cases where only relatively fine-grained sediments are analyzed, e.g., using criteria like “samples with >20% less than 63 µm” or “samples with >1% Al” as an arbitrary threshold. It is therefore not surprising to find site-specific sediment background values that span two orders of magnitude for individual metals.3
Normalization is defined here as a procedure to correct both background and contaminant concentrations for the influence of the natural variability in sediment granulometry and mineralogical composition mediated by the ambient energy of the aquatic system.4–6 It is mainly aimed at differentiating between natural variability and anthropogenic input of contaminants. Several normalization methods are commonly used, ranging from sieving, as a straightforward granulometric approach, to more complex geochemical normalization models. Whatever option is chosen, the base quality criterion is that after normalization of equally contaminated, pretreated and analyzed sediment samples, but with different grain-size distributions, the normalized concentrations should not differ significantly and should show no relation with the normalizer concentration. The benefits and drawbacks of the different approaches for achieving this criterion will be briefly discussed in the following sections to act as a guide in choosing the most appropriate approach.
2. Normalization methods
2.1. Granulometric normalization
Sedimentary components of high contaminant affinity or binding capacity for contaminants are in most cases represented by the clay fraction. Clay minerals are naturally metal-bearing and acquire additional contaminant content through highly sorptive coating formation (e.g., organic matter or Fe/Mn oxides), i.e., the natural contaminant inventory increases with the clay content. The clay content in sediments can therefore be considered as a primary normalizer. Separation of the <2 µm (9φ) clay fraction represents therefore the primary normalization method. Note, that this fraction is the only justified measure to compare metal contaminant concentrations of recent sediments directly with a shale background reference.However, separation of the clay fraction is quite laborious, and it was thus suggested to use somewhat larger mesh sizes thereby including part of the silt fraction: <16 µm (6φ),7 <20 µm (5.6φ),8 <125 µm (5φ),9 and <150 µm (4.6φ),10 with the <63 µm (4φ) fractions being that most widespread in monitoring use. In principle, the latter divides silt from sand. Note, however, that this is a purely physical property based distinction (the point at which the particle bond/weight ratio approaches unity and dry cohesive forces cease).11 Though this silt fraction, on a worldwide basis, is still mainly composed of quartz particles, positive correlations to the clay content have been frequently reported, in particular, at sites where loess occurs in the watershed. Consequently, sieving has not only been suggested as a surrogate for primary normalization of heavy metals, but also for organic contaminants.12
The international EU project on Quality Assurance of Sample Handling (QUASH) was developed as a direct response to the requirements of the Oslo & Paris Commissions (e.g., for the OSPAR Coordinated Environmental Monitoring Programme, CEMP), the Helsinki Commissions (HELCOM), and the Mediterranean Pollution and Research Programme (MEDPOL), to establish an holistic quality management and training programme to assess the effect normalization procedures have on the overall analytical uncertainty associated with the measurement of contaminants that are mandatory in national monitoring programmes. The situation is compounded by insufficient test materials and QC check samples to validate sieving methods. For the first time, QUASH provided an intercalibration exercise on sieving procedures (fractions <63 µm and <20 µm) often referred to as the weakest link in the overall analysis quality chain.13 In brief, this study has revealed that in the cases where wet sieving is applied, the between-laboratory variability in the sieving yield (sieving error) appears to be less than the respective analytical variability (analytical error), and eventually even less than the compositional variability on an individual sampling site (field error).14 For all grain-size fractions above 1% of the bulk sediment, the relative standard deviation (RSD) is usually better than the analytical target (12.5%) accepted by international QC organizations like QUASIMEME (Quality Assurance of Information in Marine Environmental Monitoring in Europe). There are thus none of the often cited limitations in the applicability of wet sieving using modern equipment.
Sediments have to be agitated during wet sieving in order to prevent clogging of the mesh and to disintegrate agglomerates. The QUASH exercise revealed no significant difference in the results between ultrasonic treatment and vibrating table agitation. For the latter, more common approach, however, a closed water system recirculated on-line by continuous-flow centrifugation, makes sieving not only less laborious but also reduces the overall water-to-solid ratio and hence any leaching or contamination effects during wet sieving. Seawater accelerates the settling of fine particles after sieving, but the fines have to be homogenized thoroughly, as centrifugation produces inhomogeneous samples.15 The centrifuged sediment subsamples should be freeze-dried and subsequently homogenized by ball milling. Air drying cannot be recommended, because the volume of air that passes over the sample during the drying process may contaminate the sample with trace organics. Polyamide sieve meshes should be checked regularly for pits created by fine-grained but sharp-edged grit. This will effectively dilute the sample with larger particles. Laboratories that seek for improvement of their actual sieving methodology may benefit from a video distributed by the QUASH Project Office (www.quasimeme.marlab.ac.uk/QUASH/quash.htm).
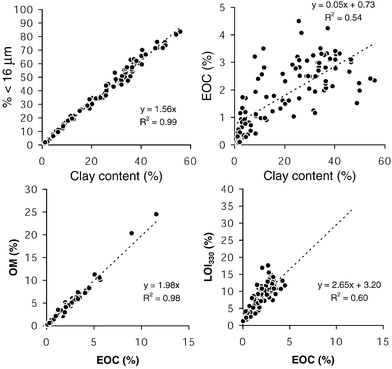
The overall practicability of sieving tends to increase with increasing mesh size, but at the cost of increasing residual variance arising from compositional (mineralogical) differences. In a related interlaboratory comparison exercise using samples from different estuaries in Europe, the sieving process was evaluated by analyzing the clay content in the fines.16 While there was a strong correlation between the clay content and the <16 µm fraction (Fig. 1), the clay content in the fraction <63 µm showed a range of a factor of four implying that sieving does not result in mineralogically equal samples. Grain-size separation does not necessarily reduce the differences between the composition of the sieved samples. Sieving, therefore, clearly cannot provide the final normalization step.13 The range of residual variation is usually small within one area, but might still be significant between different areas due to different mineralogical composition, and further geochemical normalization is then required.
 | ||
| Fig. 1 Scheme of the relationship between contaminant and potential normalizer in sediments. For the various parameters see text. | ||
2.2. Geochemical normalization
Other elements were also found to be potential grain-size proxies using correlation matrix or scatterplots, but have not been tested over broad geographical areas. In the case of pollution by titanium dioxide and bauxite processing factories, or in the case of significant amounts of Al-bearing silicates in the reach of the monitoring area (e.g., feldspars), Al or Fe cannot be used as co-factors, but Li seems to be a promising alternative proxy for the coarse fraction.26–28 Some more exotic non-anthropogenic elements like Sc, Cs, Rb and Y might then be used instead, but have been applied only in rare cases.29–31 Note also that care should be taken to apply geochemical co-factor normalization in retrospective trend monitoring based on sediment profiles where there is a hint to, but a lack of a full understanding of, post-depositional early-diagenetic processes, which can create important natural enrichments at certain sediment depths.32 Application of organic matter (e.g., TOC) as a co-factor for metal normalization in retrospective trend monitoring is also critical due to its role as a reactive component in such early-diagenetic processes.
There is no consensus yet which parameter best represents the OM content (total organic carbon, TOC; elemental organic carbon, EOC; particulate organic carbon, POC; loss-on-ignition, LOI; at different temperatures, etc.), and intercomparison exercises often yield the worst results with respect to the between-laboratory variability in OM analysis. This situation deserves a brief evaluation of the parameters currently most often used. An analysis of sediment samples from 23 European estuaries revealed that the EOC co-factor (equal to the TOC) seems to be preferable due to its much stronger correlation to OM (determined by dichromate oxidation) than the common LOI at 550 °C (LOI550) parameter. The LOI550 method has been questioned, because at temperatures above 400 °C hydroxyl-water from clay minerals is expelled, which can result in a severe overestimation of the OM content.35 From the slope, an EOC content of 48% was calculated for OM. Alternately, LOI at 330 °C (LOI330, but extended to as much as 124 h) showed a strong correlation with the EOC content (Fig. 1). EOC, OM and LOI330 showed positive linear correlations with both CB and PAH concentrations, but also an intercept deviating from zero for the latter components. In a log–log plot, the slope becomes 1 for the CBs only with LOI330, but deviates from 1 when OM or EOC is chosen as a co-factor, particularly with sandy sediments.16 For PAHs, the slope was significantly different from unity when using EOC or OM, but approached unity again when using LOI330. The latter co-factor, albeit operationally defined, appears to be an efficient parameter to characterize the OM relationships of organic contaminants. Such thorough method evaluations based on larger data sets, however, are yet to be reported.
When choosing an appropriate OM co-factor for normalization, one should bear in mind that organic chemicals may enter the aqueous environment by way of different pathways. CBs enter the aqueous environment mainly through the water or gas phase and are thus distributed by simple particle/water partitioning. PAHs, however, may also enter by particulate phases, such as charcoal and soot (also referred to as black carbon36). The latter have been recognized as a possible cause for anomalously high KOC values found in harbour sediments.37,38 Sorption of several PAHs from water to model soot particles has been measured directly only recently and was found to be 35–250 times higher than predicted, based on bulk OM.39 This indicates that EOC is not necessarily a homogeneous co-factor. Care has to be taken to identify such fine-grained but relatively supersorbent anthropogenic EOC fractions when present in significant amounts, which may obscure normalization by the OM co-factor. The key issue for normalization is thus proper characterization of the OM by as many parameters as possible, most of them rather simple and inexpensive. The types of information that can be obtained by the utilization of at least the few key parameters discussed above are often complementary and extremely useful, considering the complexity and diversity of OM encountered in the sediment environment.
3. Discussion
3.1. Normalization procedures
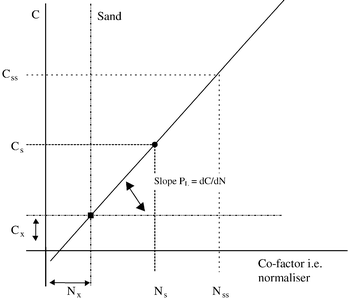
From the above discussion, it becomes clear that a two-tiered normalization approach including both sieving and co-factor measurements is to be preferred. The latter point can be rationalized by a discussion of the theoretical background of the geochemical normalization procedure. The general model for normalization based on a geochemical component mixing model is given in Fig. 2. CX and NX represent the co-factor and the contaminant contents possibly present in the coarse material (e.g., Al in feldspar or EOC in charcoal grains) and can be estimated from samples without fines. The regression line between the contaminant and co-factor will originate from that point. Regressions of co-genetic data sets but with a different contamination levels will have this point in common but tend to develop different slopes from this “turning point”. In principle, therefore, only one additional sample is required to estimate the slope for a co-genetic sample set if this turning point is known. The slope for this sample with a contaminant content CS and a co-factor content NS can be expressed as follows: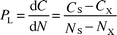 | (1) |
 | ||
| Fig. 2 Regressions between different primary normalizers analyzed in sediments from 23 representative estuaries in Europe. | ||
The clear benefit from this classical regression analysis is that an ecotoxicological quality objective (EQS) can be defined with a hypothetical reference sediment of standard composition and thus fixed content of co-factor NEQS, but with a slope and hence pollution degree equal to the measured sample to be evaluated:
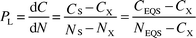 | (2) |
Normalization to a geochemical co-factor (e.g., Al or Li) level is the prerequisite for a definition of a grain-size-independent EQS, which allows for comparison of the data from total and sieved samples irrespective of the mesh size actually used. Provided there is a sufficient amount of co-factor present, an accurate result is obtained, especially with sieved samples.16 Regression lines drawn for samples from different areas may thus be used to compare their degree of contamination. The steeper the gradient, the more contaminated an area is considered to be.40,41 Positive residuals that plot above this line indicate that the concentrations are greater than would be predicted from the contaminant/co-factor relationship, and may represent hot-spot samples. However, care has to be taken to include these samples in the regression analysis (see the discussion of the statistics below). An important prerequisite for the regression approach is a sufficient co-factor variability, which is not often the case in confined monitoring areas. In the following section on criteria for quality assessment of normalization procedures, some hints are provided on how to obtain such a variability artificially, even with only a few samples.
Clearly, the contaminant/co-factor ratio approach will give anomalous results in case of non-zero intercepts. Mn, Cd, As, or other trace metals may be partitioned by early-diagenetic processes to reactive minerals such as sulfides, which are less related to the chosen co-factor. In such cases the regressions may have a significant intercept CX at a residual contaminant and/or co-factor concentration. Even negative intercepts may result, if samples with concentrations below the detection limits were treated as having a value of zero and are combined with non-zero co-factor measurements. If the coarser silicate fraction has been co-extracted, e.g., by using a total HF digestion, the CX coefficient is determined by the content of the contaminant and the geochemical co-factor in this fraction (Fig. 2). When the content of the co-factor in this fraction is NX, eqn. (2) converts to:
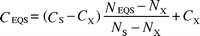 | (3) |
The latter approach may thus not apply equally well to all contaminants at all sites, even if the quality of regression with a contaminant may be striking for the individual areas based on the same elemental co-factor. The precision of the result strongly depends on the natural (or analytical) variability of NX. For coarse-grained samples, a significant standard deviation in both the CX coefficient and the slope may arise from propagation of the errors of the analytical variation due to the overall low concentrations. The CX coefficient of the regression may differ significantly from site to site, in particular, when using coarser grain size fractions. For some areas, Al contents in the coarse fractions are found at the same level as in the fines, and therefore the intercept NX becomes very high. This implies that the denominator is the result of subtracting two relatively large numbers, NS and NX. Consequently, due to their individual uncertainties, the result has an extreme error. Obviously, normalization with lower intercepts using fine-grained sediment (or sieved fines only) is more accurate.
The amount of the CX coefficient may also differ if different digestion procedures were applied. For example, when using Al as a co-factor, a higher CX coefficient may be obtained by HF than by partial nitric acid digestion, but CX coefficients derived for other co-factors such as Li may be less sensitive to the digestion method. Fig. 3 shows the correlation of the clay content with Al and Li for 23 representative estuaries in Europe, differentiated for partial (HNO3 at 140 °C) or total (HF) digestion. Strong correlations were found for both co-factors with the partial digestion method, but correlations became much weaker upon total HF digestion, with a significant increase in the intercepts. The results suggest that the aluminium content obtained by total digestion is not the best choice for normalization representing the contents of fines. For a partial digestion method, however, the intercept and slope hardly differ between areas, and correlation coefficients become even higher.
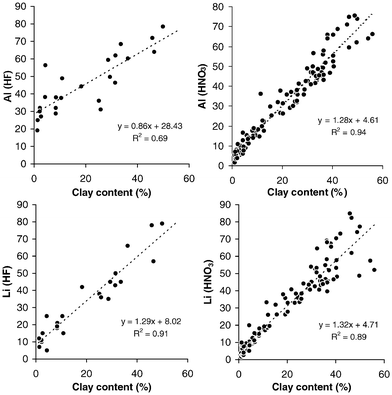 | ||
| Fig. 3 Regressions between different secondary (geochemical) normalizers analyzed in sediments from 23 representative estuaries in Europe upon partial (HNO3) or total (HF) digestion. | ||
For Li, the variability of the intercepts with different digestion methods are less significant. They are similar using both a partial and a total digestion method (Fig. 3). Moreover, using a total digestion method, there was no spatial influence on the CX coefficient as was found with aluminium. This is because Li nearly exclusively represents the clay fraction as a co-factor, whereas Al is contained also in other silicates. In general, CX coefficient variability may be more important for the whole sediment than for the fine-grained sample fractions, and due to natural variability near the intercept the representativeness for the clay content is very inaccurate for both options even if the intercept is subtracted. It is worthwhile to add, that the question of using total versus partial digestion is not merely a philosophical debate. Significant differences are often observed between partial and total digestion techniques for the co-factors, but not necessarily for metals in the sediment fines.42–44 While from a purely analytical chemists point of view, total HF digestion may be preferable due to better control of the analytical performance, for a straightforward environmental assessment this analytical paradigm has created a lot of problems, such as those discussed above.
4. Quality assessment of normalization
4.1. Geochemical criteria for quality assessment
The choice of a co-factor is justified only if its relationship with the contaminant content is equal throughout all fractions, in principle, even down to the single particle level (equilibrium assumption). A simple criterion for the applicability of a normalization procedure, e.g., for a reliable comparison of the contamination degree of samples from different sites, is that after normalization of equally contaminated, pretreated and analyzed sediment samples with very different grain-size distributions, the results should not differ significantly and should show no relation with the normalizer concentration. However, sample sets to test normalization approaches for this criterion are not always available. Sediment of different grain-size distributions may be composed from material of different origin whereby there may be no equilibrium at the particle level. Such an equilibrium can be expected only in areas where the sediment is thoroughly mixed hydrodynamically in a homogeneous water body (Fig. 4).45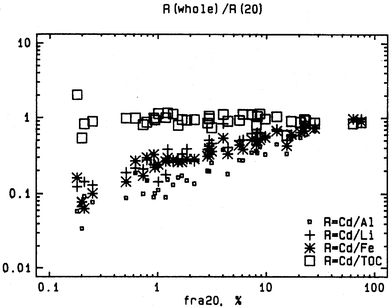 | ||
| Fig. 4 A quality criterion for a normalizer is that the contaminant/co-factor ratio R = C/N for the whole sediment should equal those in the fines, which is met here only for Cd/TOC. | ||
An alternative approach to evaluate the quality of a normalizer (a grain-size fraction, compositional co-factor or a linear combination thereof) was exemplified by the second round of the QUASH international laboratory performance assessment project.13 The new approach, as also recently demonstrated with sediments from Venice Lagoon,28 is to fractionate samples into subsamples of different grain-size distributions by wet sieving. Both the fine and coarser subsamples (e.g., >63 µm, <63 µm, 20–63 µm, and <20 µm) are then analyzed for the contaminants and co-factors in addition to the whole sample. In that way, a high variability in the co-factor concentration, i.e., a worse case than ever will occur in nature, can be obtained in the laboratory. Both the slope and the intercept (CX coefficient) can be estimated at a higher precision due to the increased regression range. Moreover, this approach provides a well equilibrated sample population for evaluation of the sensitivity of a potential geochemical co-factor. Such an evaluation could be performed, e.g., by calculation of the contaminant/co-factor ratio R = C/N [eqn. (2)] for the whole sediment and any of the grain-size fraction subsamples. The ratio in the whole sediment should then equal those in the fines, e.g., the ratio r = Rwhole/R<20 µm should equal 1. Fig. 4 shows for the example of Cd in German Bight sediments and four different co-factors that this is unfortunately not always the case. If this quality criterion is fulfilled, however, a practical consequence would be that complete separation of a fine fraction from the bulk sediment is then not necessary as, once normalized, concentrations do not vary significantly between the different sieved fractions.
4.2. Statistical data analysis
Multivariate statistical evaluation of geochemical sediment data may benefit from normalization when the grain-size distributions of sediments in a study area vary over a wide range. A significant problem with the classical regression approach, however, is the uncertainty of how to treat and interpret outliers; for example, whether positive residuals between the measured value and a selected regression (base)line reflect a degree of contamination. A significant amount of such outliers results in skewed or multi-modal data distribution frequently encountered with monitoring data. From a statistical point of view, classical parametric uni- and multivariate statistical analysis including confidence band estimation between co-factor and metal can, in principle, not be used with non-normally distributed data sets. A few samples with unusual sediment chemistry can exert a major influence on the value of the regression coefficients, in particular, for developing baseline contaminant/co-factor regression lines. A common solution to this problem involves the careful pre-examination of the raw data and removal of obvious outliers in order to reduce the skew.Exploratory data analysis (EDA) is helpful for identifying data outliers on a less subjective basis.46 It provides a number of simple graphical techniques to study the data in detail, e.g., the histogram, a one-dimensional scattergram, the box-and-whisker plot, and the, most useful, cumulative distribution diagrams. Histogram plots provide helpful delineation of the frequency of analytical data and may reveal outlier groupings (and in some, albeit not rare, cases even problems in reporting units47). Matschullat and coworkers46 discuss some robust statistical measures that can be applied to the original data set in order to identify outliers. For the next step, a number of statistical methods exist to test for the normal distribution hypothesis beyond the simple histogram plot. A large deviation between mean and median and between mean standard deviation and median absolute deviation (“mad”, a robust measure of dispersion to skew and outliers) provides another simple first indication that the data do not exhibit a normal distribution. A more reliable method of identifying outliers by a robust correlation analysis is the distance–distance (D–D) plot implemented in the statistical software package (e.g.) S-PLUS.48–50 The Shapiro–Wilk test ultimately provides the most advanced test from a statistical theory point of view to ascertain the normality hypothesis as a prerequisite for accurate least-squares regressions (LSR), either for grain-size normalization or pollution-degree evaluation.
Like other statistical methods, such as factor analysis, LSR analysis is rather sensitive to the presence of outliers, which, if present, may strongly bias the classical correlation matrix.50 An alternative approach would be application of weighted LSR, letting weight vary inversely with variance. This would cause observations of lower contaminant concentrations to receive higher weights. However, this appears to be unsatisfactory due to the concomitant decrease in overall accuracy. Tests of significance can therefore only be used as informal indicators, and this also applies to implicit significance tests such as the use of 95% prediction intervals. Using linear regression on log-transformed data has also been suggested as a way of eradicating this problem.51 However, except for the trivial case of a zero y-intercept and a slope of unity, the linearity of log-transformed data implies curvilinear relationships on linear scales, which is inconsistent with the geochemical linear mixing model. Another alternative approach, which does not require any prior knowledge about the data distribution, is to use the robust regression method of least absolute values (LAV).30 The operation of this regression method does not involve any complex weighting functions and reduces the effect of extreme values as their influence on the regression is a linear rather than a quadratic function. Moreover, LAV calculations can be carried out on the most commonly used statistical packages with only minor modifiations.30 Though the current experience with this and other robust covariation methods is rather limited, it may yield a substantially better and more reliable application of geochemical normalization than any of the classical options, if sampling involves highly contaminated and less representative locations (hot-spots) within areas.
5. Conclusions and recommendations
The aim of this review was to compare and discuss the different normalization procedures that are used in the context of monitoring contaminants in sediments. The current discussion has been stimulated by the OSPAR Commission for the Protection of the Marine Environment of the North-East Atlantic, which also provides extended guidelines for monitoring contaminants in sediments (to be found on its homepage www.ospar.org under item “Measures”). From a review of the present literature and from recent experience with the QUASH international laboratory performance studies, we derived the following recommendations on how to improve the power and comparability of sediment monitoring results by normalization.(1) The easiest approach to both temporal trend and spatial monitoring would be to analyze co-genetic samples with equal bulk composition. This could be confirmed by the determination of co-factors, Al, Li, EOC, and grain-size distribution. However, this situation is unlikely to occur, particularly with spatial surveys or even in the case that fine-grained sediment is sampled exclusively.
(2) The separation of a fine fraction from the whole sediment by wet sieving is a direct way to reduce the variance arising from grain-size differences. Spatial distribution surveys of the concentrations of contaminants in separated fine fractions can be used to prepare maps which will be much less influenced by grain-size differences than maps of whole sediment analyses. For practical reasons, the <63 µm fraction is most appropriate for both temporal trend and coordinated large-scale spatial surveys, recognizing that isolation of this fraction is an additional step to be added to the analytical procedures used in some laboratories.
(3) Upon granulometric normalization, there will likely still be some residual variance arising from differences in the composition (mineralogy and organic carbon content) of the sediments. In such a case, a two-tiered approach is to be preferred, i.e., in addition the results have to be normalized using geochemical co-factors for clay minerals or organic matter. In general, the choice of the most effective co-factor (i.e., that which results in the lowest residual variance and y-intercept) should be checked at each data assessment, by both statistical and geochemical approaches. In the case of Al being used as the (most common) co-factor, partial digestion methods (e.g., HNO3 in a microwave oven) provide a substantial decrease in the y-intercept influence. Li may be more suitable as a normalizer for metals due to the negligible y-intercept influence of coarse-grained silicate minerals. Inhomogeneity, anthropogenic impact by black carbon particles, and non-conservative behaviour of organic matter warrants site-specific evaluation of the most appropriate co-factor for organic contaminants (EOC, LOI, etc.). In principle, it is not necessary that the same normalizer be used for every survey, and also not that the same normalizer be used on each occasion that the data from any individual location is assessed, but data for the concentrations of a range of co-factors should be always made available for intercomparison purposes and, in particular, also between data that may be available for different grain-size fractions (<20 µm or <150 µm). This should ultimately give the most consistent and internationally comparable data sets over the OSPAR, HELCOM, and MEDPOL convention areas.
(4) A zero intercept facilitates enrichment factor estimations. The regression method is then also useful for evaluating any changes in contaminant load in an area if the slope is determined over regular time intervals, and may even provide a sound scientific basis to set ecological quality targets.
Assessments arising from monitoring data are critically dependent on the quality assurance of the data provided. Currently the data centre for handling environmental marine monitoring data is at the International Commission on the Exploration of the Sea, at Copenhagen (www.ices.dk). Independent scientific advice, statistical processing to enable comparisons between data sets (e.g., within CEMP), mapping, etc., are also provided. It is therefore important that the role of ICES (or similar international environmental databank centres) in this process is continuously developed and strengthened. However, in order to clarify aspects of data interpretation, a minimum of supporting analytical and statistical QA/QC information must be provided and evaluated.
Acknowledgements
This report is the output of a discussion lasting for several years by the ICES Working Group on Marine Sediments in Relation to Pollution (WGMS), chaired previously by the first and presently by the second author, which finally led to the formulation of “Sediment Guidelines” recommended for use in monitoring within the OSPAR, HELCOM, and MEDPOL convention areas. We thank all WGMS members who contributed to these discussions, in particular, Horst Albrecht, Ingemar Cato, Ian Davies, Doug Loring, Steve Rowlatt, and Birgit Schubert.References
- G. F. Birch, S. E. Taylor and C. Matthai, Environ. Pollut., 2001, 113, 357 CrossRef CAS.
- A. Helland, Water, Air, Soil Pollut., 2001, 126, 339 CrossRef CAS.
- P. M. Chapman, F. Wang, W. J. Adams and A. Green, Environ. Sci. Technol., 1999, 33, 3937 CrossRef CAS.
- D. H. Loring, ICES J. Mar. Sci., 1991, 48, 101.
- D. H. Loring and R. T. T. Rantala, Earth Sci. Rev., 1992, 32, 235 CrossRef CAS.
- K. D. Daskalakis and T. P. O'Connor, Environ. Sci. Technol., 1995, 29, 470 CAS.
- A. J. de Groot, W. Salomons and E. Allersma, in Estuarine Chemistry, ed. J. D. Burton and P. S. Liss, Academic Press, London, 1976, p. 131 Search PubMed.
- F. Ackermann, H. Bergmann and U. Schleichert, Environ. Technol. Lett., 1983, 4, 317 Search PubMed.
- A. J. Horowitz and K. A. Elrick, Appl. Geochem., 1987, 2, 437 CrossRef.
- R. Salminen, T. Tarvainen, A. Demetriades, M. Duris, F. M. Fordyce, V. Gregorauskiene, H. Kahelin, J. Kivisilla, G. Klaver, H. Klein, J. O. Larson, J. Lis, J. Locutura, K. Marsina, H. Mjartanova, C. Mouvet, P. O'Connor, L. Odor, G. Ottonello, T. Paukola, J. A. Plant, C. Reimann, O. Schermann, U. Siewers, A. Steenfelt, J. Van der Sluys, B. deVivo and L. Williams, FOREGS Geochemical Mapping Field Manual, Geological Survey of Finland, Espoo, 1998, OPAS Guide 47 Search PubMed.
- A. M. Assallay, C. D. F. Rogers, I. J. Smalley and I. F. Jefferson, Earth Sci. Rev., 1998, 45, 61 CrossRef CAS.
- J. C. Klamer, W. J. M. Hegeman and F. Smedes, Hydrobiologia, 1990, 208, 213 Search PubMed.
- F. Smedes, I. M. Davies, D. Wells, A. Allan and V. Besada, Interlaboratory Study On Sieving And Normalization Of Geographically Different Sediments, FRS Marine Laboratory, Aberdeen, Quality Assurance of Sampling and Sample Handling (QUASH) Interlaboratory Study Report WP4/R5, 2000 Search PubMed.
- B. S. Krumgalz, G. Fainshtein, M. Sahler and L. Gorfunkel, Mar. Pollut. Bull., 1989, 20, 64 CrossRef CAS.
- H. A. Van der Sloot and J. C. Duinker, Environ. Technol. Lett., 1981, 2, 511 Search PubMed.
- F. Smedes, Zand, Slib en Zeven: Standaardisering van Contaminantgehaltes in Mariene Sedimenten, RIKZ, The Hague, 1997, Report 96.043 Search PubMed.
- H. L. Windom, S. J. Schropp, F. D. Calder, J. D. Ryan, R. G. Smith, L. C. Burney, F. G. Lewis and C. H. Rawlinson, Environ. Sci. Technol., 1989, 23, 314 CAS.
- P. J. Hanson, D. W. Evans, D. R. Colby and V. S. Zdanowiecz, Mar. Environ. Res., 1993, 36, 237 CrossRef CAS.
- J. K. Summers, T. L. Wade, V. D. Engle and Z. A. Maleb, Estuaries, 1996, 19, 581 Search PubMed.
- P. W. Balls, S. Hull, B. S. Miller, J. M. Pirie and W. Proctor, Mar. Pollut. Bull., 1997, 34, 42 CrossRef CAS.
- S. Covelli and G. Fontolan, Environ. Geol., 1997, 30, 34 Search PubMed.
- S. B. Weisberg, H. T. Wilson, D. G. Heimbuch, H. L. Windom and J. K. Summers, Environ. Monit. Assess., 2000, 61, 373 CrossRef CAS.
- M. Kersten, C.-D. Garbe-Schönberg, S. Thomsen, C. Anagnostou and A. Sioulas, Environ. Sci. Technol., 1997, 31, 1295 CrossRef CAS.
- G. R. Helz and S. A. Sinex, Mar. Chem., 1986, 20, 1 CrossRef CAS.
- J. H. Rule, Environ. Geol. Water Sci., 1986, 8, 209 Search PubMed.
- D. H. Loring, Mar. Chem., 1990, 26, 155 CrossRef CAS.
- S. M. Rowlatt and D. R. Lovell, Mar. Pollut. Bull., 1990, 28, 324 CrossRef CAS.
- S. Miseroccchi, L. G. Bellucci, M. Marcaccio, G. Matteucci, V. Landuzzi, G. Quarantotto and G. Rovatti, J. Environ. Monit., 2000, 2, 529 RSC.
- F. Ackermann, Environ. Technol. Lett., 1980, 1, 518 Search PubMed.
- A. Grant and R. Middleton, Estuaries, 1998, 21, 197 Search PubMed.
- J. Prokisch, B. Kovacs, A. J. Palencsar, I. Szegvari and Z. Gyori, Environ. Geochem. Health, 2000, 22, 317 Search PubMed.
- C. Gobeil, R. W. MacDonald and B. Sundby, Geochim. Cosmochim. Acta, 1997, 61, 4647 CrossRef CAS.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, Wiley-Interscience, New York, 1993 Search PubMed.
- B. Schubert, P. Heininger and H. Bergmann, Proceedings of the Workshop "Belastung der Elbe und ihrer Nebenflüsse mit organischen Schadstoffen", ARGE Elbe, Hamburg, 1995, p. 297 Search PubMed.
- A. Goldin, Commun. Soil Sci. Plant. Anal., 1987, 18, 1111 Search PubMed.
- M. W. I. Schmidt and A. G. Noack, Global Biogeochem. Cycl., 2000, 14, 777 Search PubMed.
- Ö. Gustafsson, F. Haghseta, C. Chan, J. MacFarlane and P. M. Gschwend, Environ. Sci. Technol., 1997, 31, 203 CrossRef CAS.
- M. T. O. Jonker and F. Smedes, Environ. Sci. Technol., 2000, 34, 1620 CrossRef CAS.
- T. Bucheli and Ö. Gustafsson, Environ. Sci. Technol., 2000, 34, 5144 CrossRef CAS.
- M. Kersten and I. Kröncke, Helgoländer Meeresunters., 1992, 45, 403 Search PubMed.
- M. Fukue, T. Nakamura, Y. Kato and S. Yamasaki, Eng. Geol., 1999, 53, 131 CrossRef.
- B. S. Krumgalz and G. Fainshtein, Anal. Chim. Acta, 1989, 218, 335 CrossRef CAS.
- C. Koopmann and A. Prange, Spectrochim. Acta, Part B, 1991, 46B, 1395 CrossRef.
- G. Adami, P. Barbieri and E. Reisenhofer, Int. J. Environ. Anal. Chem., 1999, 75, 251 Search PubMed.
- H. Albrecht, ICES WGMS Report CM 1993/ENV:2, ICES, Copenhagen, 1993, pp. 28 Search PubMed.
- J. Matschullat, R. Ottenstein and C. Reimann, Environ. Geol., 2000, 39, 990 Search PubMed.
- F. T. Manheim, M. R. Buchholtz ten Brink and E. L. Mecray, J. Geochem. Explor., 1998, 64, 377 CrossRef CAS.
- W. N. Venables and B. D. Ripley, Modern Applied Statistics With S-PLUS, Springer, New York, 2nd edn, 1997 Search PubMed.
- P. J. Rousseeuw and K. Van Driessen, Technometrics, 1999, 41, 212.
- C. Reimann and P. Filzmoser, Environ. Geol., 2000, 39, 1001 Search PubMed.
- S. J. Schropp, F. G. Lewis, H. L. Windom, J. D. Ryan, F. D. Calder and L. C. Burney, Estuaries, 1990, 13, 227 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |