Reactivity of fluorinated γ-alumina and β-aluminium(III) fluoride surfaces towards hydrogen halides and tert-butyl chloride
Christopher H. Barclaya, Hamid Bozorgzadehb, Erhard Kemnitzb, Mahmood Nickkho-Amirya, Debbie E. M. Rossa, Tomaž Skapinc, James Thomson†a, Geoffrey Webba and John M. Winfield*a
aDepartment of Chemistry, University of Glasgow, Glasgow, UK G12 8QQ
bInstitute of Inorganic Chemistry, Humboldt University, Hessische Strasse 1-2, D-10115 Berlin, Germany
c“Jožef Stefan” Institute, Jamova 39, SI-1000 Ljubljana, Slovenia
First published on 29th November 2001
Abstract
The Lewis acids β-aluminium(III) fluoride and γ-alumina, fluorinated at room temperature with sulfur tetrafluoride, both interact with hydrogen fluoride and chloride, as demonstrated by radiotracer measurements using [18F] and [36Cl]. The different behaviour of HCl towards the two surfaces is rationalised by considering plausible surface sites and, in the case of β-AlF3, the role of residual water. Both materials promote dehydrochlorination of tert-butyl chloride. β-Aluminium(III) fluoride also has some catalytic activity in Friedel–Crafts alkylation whereas oligomerisation of ButCl dominates on fluorinated γ-alumina. The different behaviour appears to be due to the presence of both Lewis and Brønsted surface acidity on γ-alumina that has been fluorinated under static conditions. A description for this surface is proposed.
Introduction
It has been recognised for many years that the acidity of a γ-alumina surface can be promoted by partial fluorination of its surface1 and that this results in the formation of Al–F bonds.2 Fluorinated aluminas have been used widely as acidic catalysts or catalyst supports, for example in hydrocarbon chemistry.3 The bulk conversion of alumina to aluminium(III) fluoride, although highly favourable thermodynamically,4 is kinetically slow and it is therefore important that the materials that result from pre-fluorination processes are characterized in some detail.The situation is made more complicated by the existence of several metastable AlF3 phases in addition to the thermodynamically stable α-AlF3. The α-phase has a close-packed structure5 whereas β-AlF3 has a more open structure of the hexagonal tungsten bronze (HTB) type.6 The other phases that have been characterized structurally are related to β-AlF3.7 Although α-AlF3 has little or no catalytic activity, the β-phase is an active heterogeneous catalyst for the dismutation of chlorofluoromethanes and hydrochlorofluoromethanes.8,9 The β-, η-, θ- and κ-phases are active catalysts for the fluorination of CHCl3 or CCl3CF3 by anhydrous hydrogen fluoride.10
The catalytic behaviour of γ-alumina, pre-fluorinated using CCl2F2 or CHClF2, in C1 dismutation reactions is rather similar to that of β-AlF3. It has been suggested therefore, that in their activated form, the former materials have a surface structure that resembles that of β-AlF3.8 Although this is undoubtedly a simplification, the available evidence indicates that fluorinated alumina and the metastable AlF3 phases are closely related in their surface properties. This developing situation has been reviewed recently, emphasising the results of X-ray photoelectron studies, which are particularly informative.11 γ-Alumina, fluorinated using CCl2F2 or sulfur tetrafluoride under flow conditions, and β-AlF3 are also good heterogeneous catalysts for the isomerization of 1,1,2-trichlorotrifluoroethane to the thermodynamically preferred isomer, 1,1,1-trichlorotrifluoroethane, under flow conditions at moderate temperatures,12 suggesting that in all cases, strong Lewis acid surface sites are present.13
We now report the results of a detailed comparison of the chemical reactivity of fluorinated γ-alumina and β-AlF3 surfaces towards hydrogen fluoride, hydrogen chloride and tert-butyl chloride, which are generally regarded as very weak Lewis bases. tert-Butyl chloride was chosen as one of the probe molecules, since the catalytic behaviour of SF4-fluorinated γ-alumina and β-AlF3 in the alkylation of activated aromatics differs.14
The isolated AlF3 molecule is recognised as a very strong Lewis acid15 and species of the type AlF3(FH)n, n = 116 or 217 have been identified as vapour phase products from thermal decomposition of α-AlF3 or Al(OH)F2(H2O). From several computational studies, at various levels,18–20 of these and related species, it can be concluded that AlF3(HF) is a credible molecular species, the most recent study giving a computed value for the dissociation energy of ca. 67 kJ mol−1.20 It was therefore of interest to examine the possible interaction between HF vapour and solid β-AlF3, in which HF could be co-ordinated at surface Lewis acid sites or could be included in the hexagonal channels that are present in the structure.6 This aspect of the study was extended to determine whether there might be analogous interactions between β-AlF3 or fluorinated γ-alumina and anhydrous HCl.
We have shown previously that acid–base interactions that involve a fluorinated surface can be studied using radiolabelled probe molecules21 and that the progress of the fluorination of an oxide surface can be monitored by labelling the fluorinating agent with the short-lived isotope, fluorine-18 (t1/2 = 110 min).22,23 The high sensitivity of the radiotracer approach is a great advantage in situations where acid–base interactions are likely to be weak or difficult to observe by spectroscopic methods. A similar approach was adopted in the present work using [18F]- and [36Cl]-labelled hydrogen halides and [36Cl]-labelled tert-butyl chloride. FTIR spectroscopic measurements on SF4-fluorinated γ-alumina using pyridine as a probe molecule and mass spectrometric measurements of HCl and H2O desorption from β-AlF3 have been used to supplement the results from radiotracer experiments. Interpretations are offered for the behaviour of β-AlF3 in terms of its structure6 and the structural model previously proposed for the surfaces of β-MF3, M = Cr and Al, fluorides.11,24 The data are used also to propose a description for a fluorinated γ-alumina surface that is the result of the fluorination by SF4.
Experimental
Standard vacuum and glove box (H2O < 1 ppm) were used throughout. Except where described below, instrumentation and experimental methods have been described previously.12,25Preparation of materials
β-Aluminium(III) fluoride was prepared by the temperature programmed dehydration6 of AlF3·3H2O (5.0 g, Aldrich, purity 97%) under He (30 cm3 min−1), heating from room temperature to 493 K at 5 K min−1, held at 493 K for 1 h, heating to 723 K at 10 K min−1, held at 723 K for 2 h, finally allowed to cool to room temperature under He flow. The β-AlF3 so formed was transferred in a sealed vessel to a N2 glove box (H2O ca. 1 ppm) where subsequent manipulations were performed. Its identity was confirmed by XRD. Samples prepared in Berlin or Glasgow showed identical behaviour. Its BET area = 26 m2 g−1.12Fluorinated γ-alumina was prepared under both flow and static conditions. γ-Alumina (Degussa, BET area = 110 m2 g−1) was calcined under N2 flow at 523 K, then fluorinated under SF4/N2 flow for 2 h at 523 K (F, 47.1%; BET area = 67 m2 g−1).12 Fluorinations under static conditions were performed in a Monel metal pressure vessel (Hoke, 90 cm3) attached to a Monel vacuum line.23,25 Typically, γ-alumina (1.5 g), previously caked, sieved to produce 500–1000 μm particles and calcined in vacuo for 8 h at 523 K, was allowed to react with SF4 (9.0 mmol, 99%, Fluorochem) for 2 h, nominally at room temperature. Volatile products, a mixture of OSF2 and SO2, whose components were identified by FTIR spectroscopy, were removed by distillation and the process repeated twice. The product, an off-white solid, was transferred to and handled subsequently in, a glove box. It could be stored for short periods in FEP; storage in Pyrex led to etching, indicating that HF was lost slowly from the solid. For this reason, smaller quantities (0.5 g) were prepared for use in situ, with the appropriate adjustment of the quantity of SF4. Fluorinations were carried out also using SF4/OSF2 mixtures. Single point determinations of BET area (Coulter SA 3100 instrument) gave values in the range 80–90 m2 g−1. The imprecision was possibly a result of the corrosive nature of the material. Fluorine content was not determined directly but a value of ca. 22% was inferred from a previous [18F] study of the fluorination carried out under very similar conditions.23
Anhydrous [36Cl]-labelled hydrogen chloride was prepared from conc. aqueous HCl (10 cm3), to which was added H36Cl (1–2 cm3, specific activity ca. 925 kBq cm−3) and 98% H2SO4. Trace H2O was removed by trap to trap distillation over P2O5, the product being stored in an evacuated stainless steel vessel over P2O5.26
2-Methylpropan-2-ol (1.66 g, 23.0 mmol) was shaken with conc. aqueous HCl (5.66 cm3) containing aqueous H36Cl (1.0 cm3, 925 kBq) over a 2 h period. The lower aqueous layer was discarded and the organic layer washed with aqueous NaHCO3 then H2O.27 The tert-butyl [36Cl]chloride so formed, was dried over CaSO4 then over 3A sieves in vacuo; the yield was ca. 70%. The [36Cl] specific count rate of the vapour was 195 count min−1 kPa−1. No impurities in an inactive sample were detected using 1H, 13C NMR or FTIR spectroscopy.
Reactions under Friedel–Crafts conditions
These were performed in a Pyrex three-necked flask, equipped with a septum cap for the introduction of reagents, an He gas inlet and a gas outlet, fitted with a condenser. β-Aluminium(III) fluoride or fluorinated γ-alumina (0.5 g in each case) was loaded in the glove box and the apparatus flushed with He. A mixture of dried toluene (224.0 mmol) and dried ButCl was introduced via the septum and the mixture stirred magnetically at a constant rate to minimise diffusion effects. Samples of the liquid phase (0.5 cm3) were withdrawn at regular intervals and analysed by GC (AMS Model 93, 15 m capillary column, FID), response factors being determined by calibration with authentic samples of the products. Confirmatory measurements were made using GCMS (Hewlett Packard 5971 mass selective quadrupole detector at 70 eV interfaced to a Hewlett Packard 5890 series II GC, HP1 15 m column) and by 13C{1H} NMR spectroscopy.Radiotracer experiments
The preparation of the isotope [18F], t1/2 110 min, β+(γ) emitter, the preparation of H18F and the counting procedures used have been described elsewhere.28 Interactions between H18F and β-AlF3 or fluorinated γ-alumina (0.5 g for both solids) were studied using an evacuable Ni tube reactor equipped with a tube furnace, valves (Whitey), an FEP counting tube and a Monel vessel to contain H18F or CCl2FCClF2. The apparatus was calibrated and well seasoned with HF before use. The reactor was loaded with solid in the glove box, supporting the solid on a plug of fine Monel gauze, and transferred to a Monel vacuum line. A measured quantity of H18F (normally 1–2 mmol), whose [18F] specific count rate (values in the range 45000–8000 count min−1 [mg atom F]−1) had been determined, was introduced and the reactor heated at 573 K for 0.5–0.75 h. Uncombined H18F was removed by distillation, quantified and counted. The solid was tipped into the FEP tube and counted. After [18F] had decayed completely, the solid was weighed. Labelled β-AlF3 or F-γ-alumina (0.5 g), prepared as above, were exposed to CCl2FCClF2 (0.4–0.8 g) for 0.75–1.0 h at 523–548 K using a similar procedure.The behaviour of β-AlF3 or fluorinated γ-alumina towards [36Cl]-labelled HCl or ButCl was examined using the Geiger–Müller direct monitoring method, developed in Glasgow for [14C] adsorption measurements29 and used subsequently for a variety of inorganic applications, including those with [36Cl] and [35S].21 An evacuable Pyrex counting vessel with a gas handling facility was used for measurements at ambient temperature. Two Geiger–Müller counters were positioned to enable [36Cl] activity from the vapour phase and from the vapour plus surface (due to self-absorption of the β− emitter [36Cl], activity from the bulk was not detected) to be monitored concurrently. The counting tubes were intercalibrated using H36Cl, counts being recorded simultaneously on two scalers, enabling [36Cl] counts from the surface of a solid placed below one of the counters to be determined by subtraction. Powdered β-AlF3 or fluorinated γ-alumina (0.5 g) samples were spread as thinly as possible in order to approach the required criterion of an infinitely thin solid layer. Cell and solid were thoroughly degassed before a measured pressure of labelled H36Cl or [36Cl]-ButCl vapour was added via a calibrated gas-handling manifold. Counting times were chosen to enable substantial counts (normally 104 to minimise counting errors) to be accumulated. Pressures of volatile components were in the range 1300–6700 Pa. At the conclusion of an adsorption isotherm determination or of an addition sequence, volatile material was removed by distillation and the count from [36Cl] material retained on the solid determined.
The interaction between β-AlF3 or fluorinated γ-alumina and ButCl vapour was also studied by FTIR using a 10 cm Pyrex cell containing a depression to hold solid below the beam. It was fitted with KBr windows and an evacuable ampoule from which solid (0.5 g) could be added to the cell after a measured pressure of vapour had been added from a calibrated Pyrex vacuum manifold. The cell was supported in the spectrometer to ensure that positioning was reproducible. Spectra were recorded at regular intervals over periods up to 20 h.
Fluorination of γ-alumina with [18F]- or [35S]-labelled sulfur tetrafluoride
Samples of calcined γ-alumina (0.08, 0.10, 0.15 and 0.24 g), contained in Pyrex double limb counting vessels designed for the well of a NaI(Tl) well scintillation counter, were exposed to aliquots of SF318F (2 mmol), prepared as previously described.21 The counts developed from the solids were monitored with time. Subtraction of the very small counts determined from the vapour phase, together with the determination of the [18F] specific count rate of SF318F (as SF4,py), enabled the uptakes of [18F] to be determined. The fluorination of calcined γ-alumina at room temperature using 35SF4, which was prepared as previously described,21 was carried out in a manner similar to that described above for the [36Cl] experiments.Desorption from β-AlF3 studied by mass spectrometry
Desorption of H2O and HCl was studied using a quartz reactor with an on line coupled mass spectrometer. Before desorption was determined, the samples were heated for 1 h at 313 K in the reactor under Ar (p = 150 Pa); the system was then heated at a rate of 10 K min−1 from 313 to 673 K. The variation of the gas phase composition during constant heating was analysed using a quadrupole mass spectrometer (QMG421I Pfeiffer Vacuum GmbH). Three experiments in which β-AlF3 was treated in different ways, were performed.(a) When untreated β-AlF3 was examined, the water release was so great that no measurement was possible due to saturation of the detector. It was necessary to calcine β-AlF3in situ at 523 K, then allow the sample to cool to room temperature before following the procedure described above. (b) A fresh sample of β-AlF3 was transferred into a Schlenk tube and exposed to HCl vapour flow for 30 min at room temperature. The treated sample was transferred under inert conditions into the reactor, held at 313 K for 1 h under Ar (150 Pa) in order to remove any weakly bound HCl and heated at a constant rate as described above while desorption of HCl and H2O was monitored. (c) A β-AlF3 sample was calcined under Ar at 573 K for 2 h. After cooling to room temperature, the sample was exposed to HCl for 30 min as described above. The sample was transferred to the reactor and desorption monitored.
X-Ray photoelectron spectroscopy
The XPS measurements were performed using an ESCALAB 220 iXL spectrometer (Fisons Instruments) consisting of two vacuum chambers: the analyser and the fast entry air lock/preparation chamber. The powdered sample was fixed on carbon conductive tape (Pelco International) at the top of the sample holder and transferred into the UHV chamber. The X-ray source was monochromatic focussed Al(Kα) radiation (1486.6 eV) with an input power of 150 W. The charge on the sample was equalised with the instrument's charge compensation facility. The final peak position was determined using the C1s peak (shifted to 285.0 eV) corresponding to absorbed carbon species. The XPS-measurements were performed at a constant pass energy of 25 eV. The ESCALAB was calibrated routinely with the XPS lines of Au, Ag, and Cu.30Results and discussion
The aluminium(III) compounds
β-Aluminium(III) fluoride is a well characterized solid and was prepared by temperature programmed thermal decomposition of AlF3·3H2O, via an amorphous intermediate, AlF3·xH2O.6 γ-Alumina was fluorinated under three sets of conditions, its properties being very dependent on those used.Fluorination under flow conditions at 523 K for 2 h using a SF4/N2 gas stream12 produced a solid with F content of 47.1%. These conditions are similar to those used to fluorinate alumina with other fluorinating agents.31 Fluorination under static conditions, nominally at room temperature, was carried out using three successive additions of SF4.25 The procedure was based on a [18F] radiotracer study of the fluorination, from which an estimated value for the fluorine content was ca. 22%.23 Experiments in which γ-alumina was fluorinated in a double limb Pyrex counting vessel using a single aliquot of [18F] labelled SF4, resulted in a rapid increase in [18F] from the solid during the first 20 min and a constant value thereafter. The [18F] activity retained corresponded to an average (determined from four samples) fluorine content of 5.2%. Similar experiments using [35S]-labelled SF4 and a Geiger–Müller direct monitoring counting cell29 led to an immediate uptake of [35S] by the surface. The count rate decreased steadily over the next 0.5 h, then increased slowly over the next 0.5 h. When material that was volatile at room temperature, a mixture of OSF2 and SO2, was removed, the count rate decreased to background. Two further additions of 35SF4 to the same sample resulted in an identical pattern for the behaviour of the [35S] count rate, although the maximum value observed in situ increased from addition to addition.
Comparing this behaviour with the [18F] experiments described above and with our previous [18F] study,23 which demonstrated that the extent of the fluorination increased with each SF318F treatment, suggests the following rationalisation. An initial fluorination of the surface leads to the replacement of some surface Al–OH and Al–O–Al groups by Al–F, together with the formation of OSF2, SO2 and HF. The increasing [35S] count rates that were observed in situ over the course of the three additions of 35SF4, suggest that SO2 and possibly also OSF2, can be adsorbed weakly at the new AlIII-centred Lewis sites that have been created by the fluorination. Adsorption at basic surface fluoride sites is an alternative possibility. There was no evidence from the [35S] measurements however that sulfur-containing species were permanently retained on the surface. In contrast, the [18F] results indicated that loss of HF from the surface at room temperature was very small.
Brønsted and Lewis acidity of SF4-fluorinated γ-alumina
FTIR studies of adsorbed pyridine (py) or other basic probe molecules is a convenient method of obtaining information on the nature of surface acid sites.32 This method has been successfully applied, using photoacoustic detection, to the study of β-AlF3 and fluorinated alumina surfaces and has yielded useful information concerning the relative importance of Lewis and Brønsted sites.9 γ-Alumina, fluorinated with SF4 under flow or static conditions, was examined in this way. The spectrum of py adsorbed on the material previously fluorinated at 523 K under flow conditions, was similar to those obtained previously on CFC-fluorinated alumina.9 The relative intensities of py bands at 1452 and 1492 cm−1 indicated qualitatively that, although both types of site were present, Lewis sites predominated. Exposure of the sample to moist air followed by py treatment, resulted in the almost complete loss of IR bands associated with Lewis acidity but this could be restored by heating the sample at 423 K under N2 flow. The relative intensities of the 1452 and 1492 cm−1 bands of py adsorbed on γ-alumina that had been fluorinated at room temperature under static conditions, were reversed compared with those in the spectrum described above and indicated qualitatively that Brønsted sites predominated. This situation was particularly obvious for a sample that had been exposed to moist air.The most obvious origin of the Brønsted acidity at the surface of γ-alumina that has been fluorinated with SF4 under static conditions, is the HF that is produced from the primary fluorination of the surface by SF4. Dissociative adsorption of HF at the surface should lead to the formation of new Brønsted sites and this suggestion is consistent with the [18F] observations made above. Formation of such sites is less likely under flow conditions at higher temperature, since HF desorption will be more favoured. A second possibility is that a sulfito species was formed, due to the incomplete removal of SO2 during the surface fluorination. Some evidence for this suggestion is the observation of a peak, binding energy = ca. 167 eV and attributable to SIV, in the S(2p) X-ray photoelectron spectrum of γ-alumina that had been fluorinated under static conditions. No S(2p) peak was observed in the spectrum of material fluorinated under flow conditions. Although this second explanation appears to be inconsistent with the [35S] results reported above, very small quantities of 35SO2 retained immediately below the surface may not have been detected by Geiger–Müller counting due to self-absorption of the [35S] β− radiation. Formation of sulfito groups on or near the surface cannot be completely excluded therefore. Irrespective of its exact origin, it appears that Brønsted surface acidity is enhanced by fluorination at lower temperatures under static conditions.
Surface modification of oxidic solids, such as mesoporous silicas, by small changes in the pretreatment regime is now a well established technique.33 An example relevant to the present work is the enhancement of surface Brønsted acidity on mesoporous silica by treatment with BF3(H2O)2. Enhancement is less pronounced in silica which has been treated with BF3(OEt2).34 Exposure of both materials to py results in the observation of IR bands associated with Lewis and Brønsted acidity, the latter being more obvious when the pretreatment was with BF3(H2O)2.34
The behaviour of H18F or H36Cl towards β-AlF3 and fluorinated γ-alumina
Exposure of β-AlF3 (5.9 mmol) to H18F (1.0 mmol, specific count rate = 49482 count min−1 [mg atom F]−1) at 473 K for 1 h produced a [18F]-labelled solid. The specific count rate of the H18F recovered (0.75 mmol) was 7974 count min−1 [mg atom F]−1. This result indicated that both [18F] exchange and retention of HF by β-AlF3 had occurred. Repetition with β-AlF3 (6.2 mmol) and H18F (0.75 mmol) at 548 K for 1 h gave similar results, the proportion of HF retained being 21%. Since it is believed that HF is adsorbed on fluorinated γ-alumina (cf. above), [18F] exchange with H18F vapour should be extensive. This has been demonstrated previously at room temperature.35 The interaction between SF4-fluorinated γ-alumina and H18F at 473 K was substantial and the great extent to which H18F was retained by the solid made it impossible to quantify precisely the degree of the exchange. However, in view of the substantial [18F] exchange observed at room temperature,35 the situation at higher temperatures will be similar.Exposure of the solids that had been labelled with [18F] by this means, to CCl2FCClF2 at 548 or 523 K for 1 h did not lead to any measurable incorporation of [18F] in the organic compound. A fraction, 22%, of the H18F was lost from β-AlF3 during heating but evidently fluorination of CCl2FCClF2 did not occur. Experimental limitations due to the short t1/2 of [18F] prevented longer exposure times from being used.
The room temperature adsorption isotherm for H36Cl on γ-alumina, determined using the Geiger–Müller direct monitoring technique,29 indicated that physical adsorption and retention of a significant fraction of [36Cl] on removal of H36Cl, both occurred. This is not surprising, since we have previously shown, by [36Cl] labelling, that γ-alumina can be chlorinated under these conditions. The chlorine so deposited is strongly bound, although it is labile with respect to room temperature exchange with HCl vapour.36 Unexpectedly however, both γ-alumina, fluorinated by SF4 under static conditions, and β-AlF3 interacted at room temperature with H36Cl, albeit to a small extent. The fractions of [36Cl] surface activity retained by samples of both solids after removal of H36Cl under static vacuum are given in Table 1.
The effects on the [36Cl] surface count rates from the solids of their exposure to successive aliquots (ca. 6.6 kPa) of H36Cl, each aliquot being removed by condensation in vacuo before the next was added, are shown in Figs. 1 and 2. For the first four additions to β-AlF3 a plateau in the [36Cl] surface count rate was observed. Subsequent additions resulted in a decrease and a concomitant increase in the vapour phase count, Fig. 1. Due to the self-absorption of β− radiation emitted from [36Cl], any incorporation of H36Cl into the bulk solid would not have been detectable. Pumping the solid over several hours after the last addition removed most, but not all, of the [36Cl] activity from the surface. Using an identical procedure for fluorinated γ-alumina resulted in small but definite increases in the surface count rate over the sequence of additions, Fig. 2. Most, but not all, of the [36Cl] activity was removed on pumping at the end of the first cycle of additions. In both cases, repetition of the sequence using the same samples produced rather similar behaviour to those observed during the first series of additions.
![Variation of the [36Cl] count rate (count min−1) from the surface of β-AlF3 (■) and the vapour phase (○) with the sequential addition of H36Cl aliquots. No. 1 is the value of the surface count rate prior to the first addition of H36Cl.](/image/article/2002/DT/b106229h/b106229h-f1.gif) | ||
| Fig. 1 Variation of the [36Cl] count rate (count min−1) from the surface of β-AlF3 (■) and the vapour phase (○) with the sequential addition of H36Cl aliquots. No. 1 is the value of the surface count rate prior to the first addition of H36Cl. | ||
![Variation of the [36Cl] count rate (count min−1) from the surface of fluorinated γ-alumina (■) and the vapour phase (○) with the sequential addition of H36Cl aliquots. No. 1 is the value of the surface count rate prior to the first addition of H36Cl.](/image/article/2002/DT/b106229h/b106229h-f2.gif) | ||
| Fig. 2 Variation of the [36Cl] count rate (count min−1) from the surface of fluorinated γ-alumina (■) and the vapour phase (○) with the sequential addition of H36Cl aliquots. No. 1 is the value of the surface count rate prior to the first addition of H36Cl. | ||
The behaviour observed for β-AlF3 over the series of H36Cl additions indicates that a change in the surface occurred during the sequence. The decreasing surface and increasing vapour [36Cl] count rates observed towards the end of each series of additions, indicate that the extent of the interaction with the surface decreased. It is tempting to postulate that this is due to the incorporation of HCl in the hexagonal channels of the HTB structure. However, the ‘diameter’ of a ‘free’ channel in β-AlF3 (243 pm)6 is probably too small for HCl to be accommodated readily, making reasonable assumptions about its size (H–Cl = 127 pm, van der Waals radii of Cl and H = 180 and 120 pm respectively).37 Since the great sensitivity of radiotracer methods can result in ambiguity in interpretation when hygroscopic materials are involved, for example, due to the presence of adventitious H2O from transfer of H36Cl through Pyrex, the effect of H2O on the adsorption/desorption of HCl at β-AlF3 was studied using mass spectrometry.
The quantity of H2O evolved from uncalcined β-AlF3 on heating was too great to be measured but if the solid was calcined in situ at 523 K prior to a desorption study, evolution of H2O could be observed above 543 K. Evolution of H2O from uncalcined β-AlF3 that had been exposed to HCl flow at room temperature was observed above 373 K, Fig. 3, but no HCl was detected, Fig. 4. Treatment of β-AlF3, freshly calcined at 573 K, with HCl at room temperature led to desorption of both H2O (Fig. 3) and HCl (Fig. 4) above ca. 373 K.
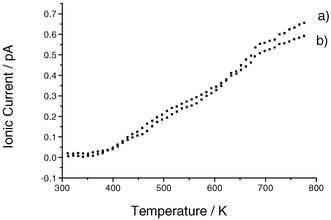 | ||
| Fig. 3 Desorption of H2O from β-AlF3; (a) freshly calcined at 573 K and subsequently treated with HCl at room temperature; (b) not calcined but treated with HCl at room temperature. | ||
 | ||
| Fig. 4 Desorption of HCl from β-AlF3; (a) freshly calcined at 573 K then treated with HCl at room temperature, (b) not calcined but treated with HCl at room temperature. | ||
Combining the radiotracer and mass spectrometric observations indicates that some H2O is retained by β-AlF3 even when it has been calcined. Partial removal of H2O from uncalcined β-AlF3 can be achieved by HCl treatment at room temperature. Adsorption of HCl on β-AlF3 is not observed unless the level of H2O is already low but, when this condition is fulfilled, adsorption is observed and some HCl is retained by calcined β-AlF3. A co-operative effect between HCl and adsorbed H2O is indicated. Their behaviour on β-AlF3 can be rationalised by considering the role of the F-terminated hexagonal channels in the solid structure6 and a plausible model for a predominant surface plane constructed by cleavage along the channel direction. This plane contains exposed, co-ordinately unsaturated AlIII sites in a fluoride environment that are expected to be strongly Lewis acidic.11,24 Both structural features are represented diagrammatically in the Scheme (I). The intermediate in the preparation of β-AlF3 is amorphous AlF3·xH2O, x < 0.5, from which pure β-AlF3 can be obtained by heating at 723 K in vacuo.6 In the present work this step was conducted under He flow, followed by calcination in vacuo at 523 K for several hours. It is proposed that residual H2O is trapped within the channels, that it is lost slowly and that this process leads to some hydration and hydroxylation of the surface (II in Scheme 1). Although surface hydrolysis can occur during prolonged exposure to moist air, for example, previous XPS work24 indicates that the surface atom ratio O2− ∶ Al3+ in β-AlF3 can be as high as 0.25, in view of the handling procedures used here, extensive hydrolysis of the surface was unlikely.
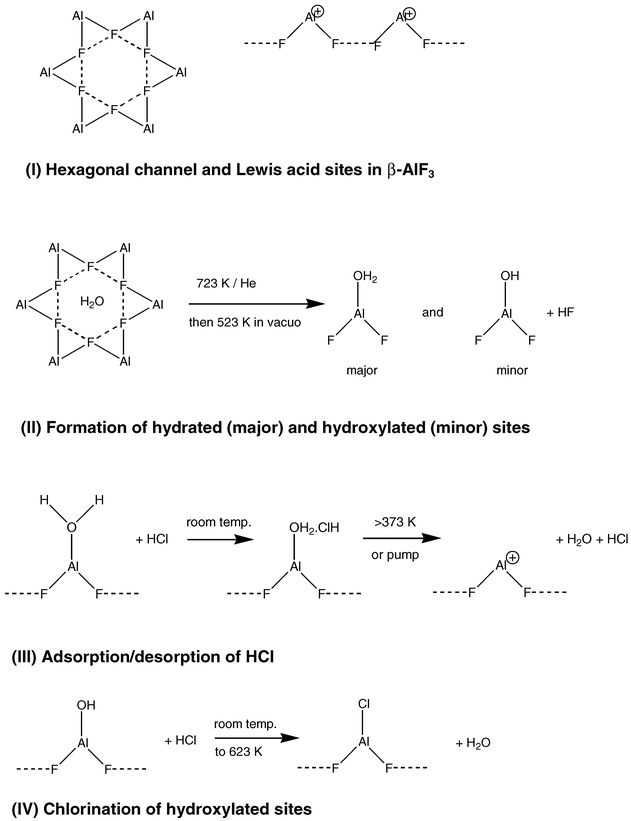 | ||
| Scheme 1 | ||
Adsorption of HCl on β-AlF3 is visualised as HCl becoming hydrogen bonded to co-ordinated H2O (III, Scheme 1). A computational study of the isolated complexes, AlF3(H2O)n, n = 2 or 3, in which one H2O molecule is directly co-ordinated to AlIII; the remainder being involved in O–H—O and O–H—F bonding,38 is a possible precedent for this suggestion. Most, but not all, of the HCl is lost under vacuum at room temperature (Table 1) and desorption of HCl and H2O are both observed by mass spectrometry above 373 K (Figs. 3 and 4). In principle, coordinatively unsaturated AlIII sites are generated by this sequence (III, Scheme 1). The decreasing [36Cl] surface count rates observed over the latter part the sequence of H36Cl additions (Fig. 1) is consistent with the decreasing hydration of the surface, if the proposal for the adsorbed state is accepted.
A very small fraction of HCl was retained on the surface after pumping for several hours (ca. 2–3% of the saturation [36Cl] surface count rate from GM monitoring). This suggests that chlorination of the surface OH groups can occur (IV, Scheme 1) in addition to weak adsorption, when β-AlF3 is exposed to HCl.
The increasing incorporation of [36Cl] on the fluorinated γ-alumina surface with repeated exposure of the surface to aliquots of H36Cl, Fig. 2, might be the result of the chlorination of unfluorinated surface hydroxyl groups. Although this may occur to a small extent, cf. the situation for β-AlF3 (Scheme 1), by analogy with the situation for unfluorinated γ-alumina,36 it would be expected that a substantial proportion of [36Cl] activity would be retained on the surface. This is contrary to the experimental findings that ca. 10% was retained when H36Cl was removed under static vacuum (Table 1) and that only 2–3% was retained after pumping over several hours. A more speculative rationalisation is that the adsorbed H36Cl is weakly hydrogen bonded to surface fluoride. This would account for the build up of [36Cl] activity observed throughout the sequence of experiments. The hydrogen bonded dimer, HF—HCl, is known to exist in the vapour phase39 but is only weakly bound.39,40 Its existence on a surface is therefore problematic. Estimated values of X−—HY, X and Y = F and Cl, dissociation energies, derived from gas phase ion-molecule reactions studied by mass spectrometry,41,42 are greater than the corresponding values for the neutral dimers.40 Dissociation energies estimated for F−—HY are 161 (Y = F) and 250 kJ mol−1 (Y = Cl).42 Therefore the formation of surface species of the type (Al)–Fδ−—HCl is more plausible.
The uptakes of HF by β-AlF3 and fluorinated γ-alumina are significantly greater than their HCl counterparts. However, the proposals made above are relevant to these systems also with two additional considerations. Incorporation of HF in the channels of β-AlF3 is a possibility and the presence of oligomers on the surface cannot be discounted.
The behaviour of tert-butyl chloride on β-AlF3 and fluorinated γ-alumina
The Lewis acid properties of β-AlF3 and fluorinated γ-alumina make them candidates for Friedel–Crafts catalysts, being possible replacements for aluminium(III) chloride in this respect. Although preliminary results indicated that both β-AlF3 and SF4-fluorinated γ-alumina demonstrated some activity in the room temperature alkylation of toluene by tert-butyl chloride,14 a detailed examination of fluorinated γ-alumina revealed a more complex situation. Unlike the situation for CCl2FCClF2 isomerisation,12 there were significant differences between the behaviour of ButCl towards β-AlF3 and that towards γ-alumina, fluorinated using pure SF4 under static conditions.A stirred mixture of ButCl and toluene (1 ∶ 10 mol ratio) reacted at room temperature in the presence of solid β-AlF3 to give a mixture of mono-alkylated products (conversion 46%, para ∶ meta = 84 ∶ 16) within 10 min, although further conversion was not observed thereafter. The behaviour of γ-alumina, fluorinated using a mixture of SF4 and OSF2 under static conditions, was similar, the conversion being 36% after 50 min (para ∶ meta = 97 ∶ 3). Alkylation of benzene under identical conditions produced small initial conversions to mono- and di-alkylated products; conversions were 5 and 8% respectively for β-AlF3 and fluorinated γ-alumina and the mono ∶ di product ratios were 79 ∶ 21 and 89 ∶ 11. In all cases unchanged ButCl was present.
γ-Alumina fluorinated by pure SF4 had no Friedel–Crafts activity at room temperature, although consumption of ButCl was significant (55–60% over 1 h). The decrease in solution concentration was particularly marked, 37%, after 5 min. The same phenomenon was observed in the absence of hydrocarbon. Under these conditions, consumption of ButCl was accompanied by the appearance of an orange layer on the solid whose colour was discharged on the admission of moist air. Analysis of the liquid reaction mixture by GCMS indicated that two non-chlorine containing species were present, tentatively identified as (CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, the obvious dehydrochlorination product from ButCl, and a C16 hydrocarbon, whose mass corresponded to a tetramer derived from the olefin but whose structure was undetermined.
CH2, the obvious dehydrochlorination product from ButCl, and a C16 hydrocarbon, whose mass corresponded to a tetramer derived from the olefin but whose structure was undetermined.
Consumption of ButCl by SF4-fluorinated γ-alumina was rapid also under solid-gas conditions at room temperature, as indicated by FTIR and [36Cl] tracer studies. The solid became coated with a yellow material. There was no evidence from FTIR for the evolution of HCl into the vapour phase, although with the pressure of ButCl used, it should have been readily detectable. Similarly, when SF4-fluorinated γ-alumina was exposed to [36Cl]-ButCl, there was an initial, substantial decrease in the [36Cl] count rate detected from the gas phase, followed by a small, continuous decrease thereafter. However, no count rate from the surface was observed. The surface changed colour during exposure from colourless → yellow → purple.
Exposure of β-AlF3 to ButCl under identical conditions resulted in its incomplete consumption and the evolution of some HCl. However, [36Cl] monitoring of the surface provided no evidence for a substantive interaction involving [36Cl]-labelled species. The behaviour of γ-alumina, fluorinated using a SF4/OSF2 mixture, was similar to that of β-AlF3, although the surface of the solid became purple with time.
γ-Alumina fluorinated using SF4 is known to promote dehydrochlorination in chloroalkanes25,35 and therefore dehydrochlorination of ButCl in the presence of these solids is not surprising. The distinctive behaviour of SF4-fluorinated γ-alumina towards ButCl can be rationalised by assuming that (CH3)2CCH2, formed by the initial dehydrochlorination reaction, undergoes rapid oligomerisation. The absence of detectable [36Cl] from the surface suggests that the organic layer so formed covers HCl adsorbed on the surface, preventing its observation by [36Cl] monitoring and its escape to the vapour phase. In this system therefore, the dehydrochlorination–oligomerisation route suppresses Friedel–Crafts activity, in contrast to the situation with β-AlF3 or γ-alumina fluorinated using an SF4/OSF2
mixture. It is concluded that the different behaviour is the result of the combination of very strong Lewis surface sites and significant Brønsted acidity on SF4-fluorinated γ-alumina.
Conclusion: the nature of the SF4-fluorinated γ-alumina surface
The similarity in catalytic behaviour found for β-AlF3 and SF4-fluorinated γ-alumina surfaces towards CCl2FCClF2 and other chlorofluoroethanes, is the result of their Lewis acidity. In catalytic situations where the presence of Lewis sites on the surface is the only factor of importance, very similar behaviour for the two materials is to be expected, as has been observed.12The behaviour of H18F and H36Cl towards β-AlF3 and fluorinated γ-alumina is formally similar and in both cases unexpected. It has, however, a different origin. For β-AlF3 the phenomenon can be rationalised in terms of the structure of the bulk6 and the proposed model for the surface of β-AlF3,11,24 however the situation for fluorinated γ-alumina is not so straightforward.
The characteristic feature of fluorinated γ-alumina is that Brønsted and Lewis acidity can both be important. The nature of the surface formed is highly dependent on the exact conditions used for its preparation. Fluorination using SF4 under static conditions and at lower temperature, results in a material with a lower fluorine content but one in which Brønsted and Lewis acidity are both manifest. This is rationalised as a result of incomplete removal of HF when fluorination is conducted under static conditions and this is responsible for the ability of the material to interact further with HF and to interact with HCl. It seems likely also that Brønsted acidity is a factor in accounting for the different behaviour of SF4-fluorinated γ-alumina compared with β-AlF3 towards ButCl.
γ-Alumina has a defect (tetrahedral AlIII) spinel structure whose stoichiometry only approximates to Al2O3 and whose surface is stabilised by the presence of hydroxyl groups.43 It is well established that surface hydroxyls on γ-alumina exist in several different environments and have, as a consequence, different acidities.44 They have an indirect influence on coordinatively unsaturated AlIII Lewis acid sites, since three types of Lewis site can be correlated with different types of –OH that are their nearest neighbours.45 Fluorination of the surface with SF4, nominally at room temperature, will result in partial replacement of Al–OH groups by Al–F and the formation of OSF2, SO2 and HF. Under static conditions HF can be adsorbed dissociatively to form F–Al–(OH)–Al groups which can potentially function as Brønsted sites and sites at which HCl adsorption or further HF adsorption can occur. Under flow conditions above room temperature, most if not all, of the HF formed is expected to be lost from the surface, therefore the formation of new Brønsted sites will be relatively less important. In this situation, Lewis acidity predominates. The surface properties are similar to those that result from fluorination with a chlorofluorocarbon or a hydrochlorofluorocarbon.31 Enhanced Lewis acidity is the result of the replacement of surface oxygen atoms by fluorine (O ≡ 2F) resulting in surface AlIII atoms which have a disordered O/F environment. New, strong Lewis sites are created with the inevitable disruption of the surface structure. It could be argued that the surface that results from room temperature fluorination would be more disordered and so have stronger Lewis sites. However, we have no direct information on this point.
Ab initio calculations, at the SV-321G level, on small clusters that are relevant to γ-alumina and its chlorinated analogues suggest that both Brønsted and Lewis acid character are associated with AlIII atoms occupying tetrahedral rather than octahedral sites.46 Replacement of OH groups by Cl, up to two Cl atoms per Al–O–Al cluster, results in significant increases in both types of acidity. In some respects, γ-alumina which has been fluorinated by SF4 under static conditions resembles material that has been chlorinated using CCl4,36 although, unlike the material fluorinated under static conditions, chlorinated γ-alumina is an efficient Friedel–Crafts catalyst at room temperature.14 The two materials also differ in the extent to which halogenation occurs and in the extent of the interactions that involve hydrogen halide. Notwithstanding these differences in properties however, Brønsted and Lewis acidity in SF4-fluorinated γ-alumina is more likely to be associated with tetrahedral AlIII sites.
Acknowledgements
We thank staff at the John Mallard Scottish PET Centre for assistance with [18F] preparation and the EU (contract ENV4-CT97–0601) and EPSRC for support of this work.References
- e.g. L. González Tejuca, C. H. Rochester, A. López Agudo and J. L. Garcia Fierro, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2543 Search PubMed; A. Corma, V. Fornés and E. Ortega, J. Catal., 1985, 92, 284 RSC.
- J. R. Schlup and R. W. Vaughan, J. Catal., 1986, 99, 304 CrossRef CAS.
- e.g. V. M. Allenger, C. Fairbridge, D. D. McLean and M. Ternan, J. Catal., 1987, 105, 71 Search PubMed; A. Corma, V. Fornés and F. Melo, Appl. Catal., 1990, 61, 175 CrossRef CAS; V. M. Allenger, D. D. McLean and M. Ternan, J. Catal., 1991, 131, 305 CrossRef CAS.
- E. Kemnitz and D.-H. Menz, Prog. Solid State Chem., 1998, 26, 97 CrossRef CAS.
- R. Hoppe and D. Kissel, J. Fluorine Chem., 1984, 24, 327 CrossRef CAS.
- A. Le Bail, C. Jacoboni, M. Leblanc, R. De Pape, H. Duroy and J. L. Fourquet, J. Solid State Chem., 1988, 77, 96 CrossRef CAS.
- U. Bentrup, Eur. J. Solid State Inorg. Chem., 1992, 29, 51 CAS; N. Herron, D. L. Thorn, R. L. Harlow and F. Davidson, J. Am. Chem. Soc., 1993, 115, 3028 CrossRef CAS; N. Herron, R. L. Harlow and D. L. Thorn, Inorg. Chem., 1993, 32, 2985 CrossRef CAS; N. Herron, D. L. Thorn, R. L. Harlow, G. A. Jones, J. B. Parise, J. A. Fernandez-Baca and T. Vogt, Chem. Mater., 1995, 7, 75 CrossRef CAS; C. Alonso, A. Morato, F. Medina, F. Guirado, Y. Cesteros, P. Salagre, J. E. Sueiras, R. Terrado and A. Giralt, Chem. Mater., 2000, 12, 1148 CrossRef CAS.
- A. Hess, E. Kemnitz, A. Lippitz, W. E. S. Unger and D.-H. Menz, J. Catal., 1994, 148, 270 CrossRef.
- A. Hess and E. Kemnitz, J. Catal., 1994, 149, 449 CrossRef CAS.
- N. Herron and W. E. Farneth, Adv. Mater., 1996, 8, 959 CrossRef CAS.
- E. Kemnitz and J. M. Winfield, in Advanced Inorganic Fluorides, eds. T. Nakajima, B. Žemva and A. Tressaud, Elsevier, Lausanne, 2000, ch. 12, pp. 367–401 Search PubMed.
- H. Bozorgzadeh, E. Kemnitz, M. Nickkho-Amiry, T. Skapin and J. M. Winfield, J. Fluorine Chem., 2001, 107, 45 CrossRef CAS.
- D. G. McBeth, M. M. McGeough, G. Webb, J. M. Winfield, A. McCulloch and N. Winterton, Green Chem., 2000, 2, 15 RSC.
- C. H. Barclay and J. M. Winfield, in Supported Reagents and Catalysts in Chemistry, eds. B. K. Hodnett, A. P. Kybett, J. H. Clark and K. Smith, RSC Special Publication No. 216, Cambridge, 1998, pp. 60–65 Search PubMed.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151 CrossRef CAS.
- D. Menz, L. Kolditz, K. Heide, C. Schmidt, Ch. Kunert, Ch. Mensing, H. G. v. Schnering and W. Hönle, Z. Anorg. Allg. Chem., 1987, 551, 231 CrossRef CAS.
- D.-H. Menz, Ch. Mensing, W. Hönle and H. E. v. Schnering, Z. Anorg. Allg. Chem., 1992, 611, 107 CrossRef CAS.
- C. Nieke, G. Scholz and D.-H. Menz, J. Fluorine Chem., 1992, 59, 41 CrossRef CAS.
- L. A. Curtiss and G. Scholz, Chem. Phys. Lett., 1993, 205, 550 CrossRef CAS.
- G. Scholz and R. Stösser, J. Fluorine Chem., 1997, 86, 131 CrossRef CAS.
- K. W. Dixon and J. M. Winfield, J. Chem. Soc., Dalton Trans., 1989, 937 RSC; T. Baird, A. Bendada, G. Webb and J. M. Winfield, J. Mater. Chem., 1991, 1, 1071 RSC; T. Baird, A. Bendada, G. Webb and J. M. Winfield, J. Fluorine Chem., 1994, 66, 117 CrossRef CAS.
- J. Kijowski, G. Webb and J. M. Winfield, Appl. Catal., 1986, 27, 181 CrossRef CAS.
- A. Bendada, G. Webb and J. M. Winfield, Eur. J. Solid State Inorg. Chem., 1996, 33, 907 CAS.
- E. Kemnitz, A. Kohne, I. Grohmann, A. Lippitz and W. E. S. Unger, J. Catal., 1996, 159, 270 CrossRef CAS.
- J. Thomson, G. Webb, J. M. Winfield, D. Bonniface, C. Shortman and N. Winterton, Appl. Catal. A: General, 1993, 97, 67 CrossRef CAS.
- L. Rowley, G. Webb, J. M. Winfield and A. McCulloch, Appl. Catal., 1989, 52, 69 CrossRef CAS.
- A. I. Vogel, Textbook of Practical Organic Chemistry, 4th edn., Longman, London, 1978, p. 383 Search PubMed.
- A. W. Baker, D. Bonniface, T. M. Klapötke, I. Nicol, J. D. Scott, W. D. S. Scott, R. R. Spence, M. J. Watson, G. Webb and J. M. Winfield, J. Fluorine Chem., 2000, 102, 279 CrossRef CAS; C. P. Kealey, T. M. Klapötke, D. W. McComb, M. I. Robertson and J. M. Winfield, J. Mater. Chem., 2001, 11, 879 RSC.
- A. S. Al-Ammar and G. Webb, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 195 RSC.
- M. T. Anthony and M. P. Seah, Surf. Interface Anal., 1984, 6, 95 CAS.
- E. Kemnitz and A. Hess, J. Prakt. Chem./Chem. Ztg., 1992, 334, 591 Search PubMed; J. Thomson, J. Chem. Soc., Faraday Trans. 1, 1994, 90, 3585 Search PubMed; T. Skapin, J. Mater. Chem., 1995, 5, 1215 RSC.
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
- P. M. Price, J. H. Clark and D. J. Macquarrie, J. Chem. Soc., Dalton Trans., 2000, 101 RSC.
- K. Wilson and J. H. Clark, Chem. Commun., 1998, 2135 RSC.
- A. Bendada, D. W. Bonniface, F. McMonagle, R. Marshall, C. Shortman, R. R. Spence, J. Thomson, G. Webb, J. M. Winfield and N. Winterton, Chem. Commun., 1996, 1947 RSC.
- J. Thomson, G. Webb and J. M. Winfield, J. Mol. Catal., 1991, 67, 117 CrossRef CAS.
- See for example: C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education, Harlow, 2001 Search PubMed.
- M. Krossner, G. Scholz and R. Stösser, J. Phys. Chem. A, 1997, 101, 1555 CrossRef CAS.
- K. C. Janda, J. M. Steed, S. E. Novick and W. Klemperer, J. Chem. Phys., 1977, 67, 5162 CrossRef CAS; G. T. Fraser and A. S. Pine, J. Chem. Phys., 1989, 91, 637 CrossRef CAS.
- P. Kollman, J. Am. Chem. Soc., 1977, 99, 4875 CrossRef CAS.
- J. W. Larson and T. B. McMahon, J. Am. Chem. Soc., 1983, 105, 2944 CrossRef CAS.
- G. Caldwell and P. Kebarle, Can. J. Chem., 1985, 63, 1399 CAS.
- H. Jagodszinski and H. Saalfeld, Z. Kristallogr., 1958, 110, 197 Search PubMed; B. C. Lippens, Thesis, Delft University of Technology, The Netherlands, 1961 Search PubMed; S. Soled, J. Catal., 1983, 81, 252 Search PubMed.
- C. Morterra and G. Magnacca, Catal. Today, 1996, 27, 497 CrossRef CAS and references therein.
- X. Liu and R. E. Truitt, J. Am. Chem. Soc., 1997, 119, 9856 CrossRef CAS.
- J. Thomson, G. Webb, B. Webster and J. M. Winfield, J. Chem. Soc., Faraday Trans., 1995, 91, 155 RSC.
Footnote |
| † Present address: Department of Chemistry, University of Dundee, Dundee, UK DD1 4HN. |
| This journal is © The Royal Society of Chemistry 2002 |