Anionic metal–organic and cationic organic layer alternation in the coordination polymers [{M(BTEC)(OH2)4}·{C4H12N2}·4H2O]n (M = Co, Ni, and Zn; BTEC = 1,2,4,5-benzenetetracarboxylate)
Ramaswamy
Murugavel
*,
Divya
Krishnamurthy
and
Malaichamy
Sathiyendiran
Department of Chemistry, Indian Institute of Technology-Bombay, Powai, Mumbai-400 076, India
First published on 29th November 2001
Abstract
New coordination polymers [{M(BTEC)(OH2)4}·{C4H12N2}·4H2O]n (BTEC = 1,2,4,5-benzenetetracarboxylate) (M = Co 1; Ni 2; Zn 3) have been synthesized starting from the respective transition metal salts, 1,2,4,5-benzenetetracarboxylic acid and piperazine hexahydrate. The highly-crystalline compounds, which are insoluble in water as well as common organic solvents, have been characterized in the solid-state with the aid of elemental analysis, IR and diffuse reflectance UV-visible spectroscopy, and TGA/DTA measurements. The molecular structures of all the compounds have been determined in the solid-state by X-ray diffraction studies; all the three compounds are isomorphous and crystallize in centerosymmetric triclinic space group. The molecular structures are made up of extensively hydrogen-bonded alternating layers of anionic {M(BTEC)(OH2)4}n coordination polymer and piperazinium dications. The extended structure formed in the solid-state due to extensive inter-layer O–H⋯O and N–H⋯O hydrogen bonds incorporate four water molecules per unit cell. The water molecules present in the polymeric network can be easily removed at fairly low temperatures. Heating 1 or 2 to approximately 60–70 °C in a vacuum (0.1 mmHg) for 12 h results in the elimination of three water molecules as evidenced by elemental analysis. Similar results were obtained from the thermogravimetric studies. The dehydration process is accompanied by a colour change in the case of compounds 1 and 2. Exposure of the dehydrated sample to ammonia results in the uptake of three equivalents of ammonia molecules. However, the methane gas adsorption studies carried out on dehydrated sample did not show the presence of a porous structure.
Introduction
There has been a recent upsurge in the synthesis of coordination polymers1–17 which are based on multifunctional ligands because of the ability of some of these polymeric materials to form open framework metal–organic microporous materials.3,4,7c In particular, recent studies have shown the use of divalent transition metal ions and benzene di-, and tri-carboxylic acids (Chart 1) as modular precursors in the design of numerous metal–organic polymeric solids with desired topologies. Especially interesting are the complexes formed by H2BDC and H3BTC having one-dimensional polymeric chain or brick-wall structures and their selective guest binding abilities.18–24 | ||
| Chart 1 | ||
In spite of the rich coordination chemistry exhibited by H2BDC and H3BTC in the presence of auxiliary ligands and coordinated solvents, barring a few sporadic reports, which mainly concentrate on the direct interaction between the metal ion and the ligand, there have been no serious attempts to prepare metal–organic polymeric or supramolecular structures based on 1,2,4,5-benzene tetracarboxylic acid (H4BTEC).25–28 It may be argued that, due to steric reasons, all the four carboxyl groups of H4BTEC are unlikely to take part in coordination to the metal. However, even the presence of free –COOH groups (especially in the vicinity of coordinated water molecules, donor solvents and added amines) would lead to formation of new extended structures aided by hydrogen bonding interactions. Moreover, the presence of a large number of uncoordinated water molecules within the lattice, may in turn lead to interesting possibilities for the preparation of porous solids.19a
Keeping in mind the aforementioned points, we have attempted to study the extended solid forming ability of H4BTEC towards divalent first row transition metal ions, such as Co2+, Ni2+ and Zn2+ in the presence of a donor amine. Our results describing the ability of H4BTEC to form extended inorganic solids, incorporating piperazine and water molecules between the coordination polymer layers, are described in this paper.
Results and discussion
Synthesis and spectral properties
The title compounds [{M(BTEC)(OH2)4}·{C4H12N2}·4H2O]n (M = Co 1; Ni 2; Zn 3; C4H10N2 = piperazine) are obtained in good yield by the slow reaction of an aqueous solution of the respective transition metal salts, MX2·xH2O (for M = Co or Ni: X = Cl, x = 6; for M = Zn: X2 = SO4, x = 7), with H4BTEC and piperazine hexahydrate (Scheme 1). Compounds 1–3 are obtained as well formed crystals directly from the reaction mixtures. These crystals, whose size is dependent on the rate of crystallization and the concentration of the initial solution, are insoluble in water as well as common organic solvents.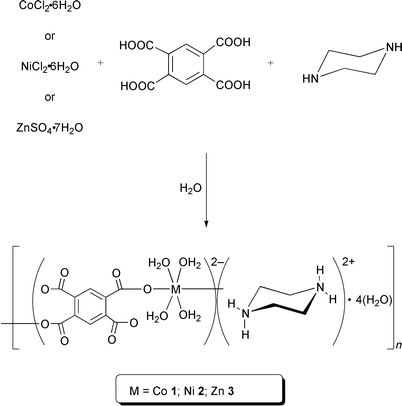 | ||
| Scheme 1 Synthesis of 1–3. | ||
The compounds show an identical infrared spectral pattern with the absorption bands of the symmetric and asymmetric carboxylate stretching vibrations appearing between 1630–1605 cm−1 and 1492–1360 cm−1, respectively. The difference between the νasym(COO−) and νsym(COO−) in the IR spectra indicate the unidentate terminal mode of coordination of the carboxylate groups.19a,29
The diffuse reflectance UV-visible spectrum of the cobalt coordination polymer 1 shows three absorption maxima in the visible region at 647, 503, and 360 nm (Fig. 1a). While the strong absorption at 503 nm is easily assignable to the 4T1g(F) → 4T1g(P), the weak absorption at 647 nm could be ascribed to the 4T1g(F) → 4A2g(F) metal d–d transition. Similarly, two absorption maxima in the visible region were observed for the nickel polymer 2 at 662 and 394 nm corresponding to the 3A2g → 3T1g(F) and 3A2g → 3T1g(P) transitions, respectively.30
 | ||
| Fig. 1 Diffuse reflectance UV-visible spectra of (a) as synthesized 1 and (b) dehydrated sample of 1, showing Oh and Td coordination environments around the cobalt ion, respectively. | ||
Thermal analysis
TGA curves of the cobalt and nickel derivatives show a stepwise loss of water molecules. The first weight loss occurring at 50–90 °C corresponds to the loss of two water molecules (possibly the uncoordinated ones) while a further weight loss observed at 100–170 °C could be attributed to the loss of two more uncoordinated and two coordinated water molecules. Independent heating of the pink coloured cobalt coordination polymer 1 at around 115 °C for 1 h results in a change of colour to blue. Exposing the dehydrated sample to moist air or soaking it in water results in the complex reverting back to its original octahedral structure. A similar behaviour was observed for the nickel coordination polymer 2, where the original green-coloured compound undergoes a colour change to greenish-yellow on heating up to 150 °C. The greenish-yellow sample reverts back to a green colour on exposure to moist air, demonstrating that the coordinated water molecules can be reversibly dehydrated.Dehydration and ammonia insertion studies
It has been well documented in the literature that coordination polymers containing a large number of water molecules can be transformed into porous solids which can serve as hosts for a variety of guest molecules.19a The results obtained from the TGA and DTA curves (vide supra) prompted us to investigate the possibility of generating metal–organic porous solids (zeotypes) from 1–3 by heat treating them at low-temperatures under reduced pressure, and use these pores to trap substrates of appropriate size and shape.To achieve this, crystalline samples of 1 and 2 were heated at ≈60 °C (0.1 mmHg; 12 h). As described above, there was a colour change in each case (pink to blue for 1, and green to yellowish-green for 2). This indicates a change in the coordination environment around the metal ions from Oh to Td, as evidenced by visible spectral measurements on the resultant sample of 1 (Fig. 1). The microanalyses of the resultant solids reveal that the amount of water lost approximately corresponds to three molecules of H2O per formula unit. Although the origin of the three water molecules that are lost cannot be unambiguously assigned, the above data points to the possible loss of two water molecules from the coordination sphere.
The IR spectra of the as prepared sample 2 and its dehydrated form are depicted in Fig. 2. It is interesting to note that the N–H absorption of the piperazinium cation is well resolved in the dehydrated sample and appears as a sharp doublet (Fig. 2b). On the other hand a very broad strong absorption between 2800–3400 cm−1, for both the OH and NH absorption is observed for the parent compound (Fig. 2a).
 | ||
| Fig. 2 IR spectra of (a) as synthesized Ni-polymer 2, and (b) a dehydrated sample (60 °C, 12 h, 0.1 mmHg) of 2 recorded as KBr plates. | ||
Assuming that the above dehydration experiment has resulted in a porous solid, ammonia (which has a similar molecular volume and shape to water) was chosen for guest inclusion studies. In a typical experiment, a large excess of ammonia gas was condensed into a Schlenk flask containing the dehydrated sample at −50 °C. The resultant mixture was kept aside for 60 min with occasional stirring during which time the excess NH3 was allowed to evaporate. Elemental analysis of the resultant solid, after air-drying, revealed the incorporation of three equivalents of ammonia per formula unit. The ammonia-incorporated samples on exposure to moist-air, even after several days, did not show any compositional changes. Further, thermal analyses of these samples showed a considerable increase in the onset of the first weight loss compared to the parent samples 1 and 2. For example, while the DTA curve of 1 shows endotherms at 84 and 138 °C for the two successive weight-losses of water molecules, the endotherms occur at 132 and 163 °C for the corresponding ammonia-impregnated sample (Fig. 3a). A similar behaviour was also observed for the Ni complex 2, whose endotherms appear at 78 and 160 °C. For the corresponding ammonia incorporated Ni derivative the endotherms are observed at 130 and 189 °C (Fig. 3b). Although these observations clearly indicate that the NH3 molecules are more tightly bound than the water molecules in the parent derivatives 1 and 2, it is not clear whether the NH3 molecules are coordinated to the metal. As a final check, to test whether the dehydration leads to a porous solid retaining the original framework structure, we carried out powder XRD (Fig. 4) and gas adsorption studies of the dehydrated samples.
 | ||
| Fig. 3 (a) DTA curves of (i) as synthesized 1 and (ii) ammonia incorporated sample of 1. (b) Corresponding DTA curves of 2. | ||
 | ||
| Fig. 4 Powder XRD pattern of (a) as synthesized, (b) dehydrated, and (c) ammonia incorporated samples of 1. | ||
The powder XRD pattern of the dehydrated sample shows that the original framework structure is retained even after dehydration (Fig. 4b). However the XRD pattern of the NH3 soaked sample shows the collapse of the original framework (Fig. 4c), indicating a possible change in the structure after the introduction of the NH3 molecules. On the other hand, gas adsorption experiments carried out on the dehydrated cobalt derivative 1 to incorporate neutral gas molecules such as CH4 (instead of NH3) did not show the expected Type I isotherm. Hence these coordination polymers cannot be strictly termed as porous solids, since they neither retain the framework structure when exposed to ammonia nor incorporate neutral gaseous molecules such as methane.
Molecular structures
In order to understand the structures of these coordination polymers in the solid-state, single crystal X-ray diffraction studies were carried out for each compound. However, the molecular structures of all three compounds were found to be isomorphous and they crystallize in the triclinic centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . A perspective view of the repeating unit of the nickel polymer 2 is depicted in Fig. 5; selected structural parameters such as bond distances and angles for 1–3 are listed in Table 1. The various types of hydrogen bonds present in these coordination polymeric networks are listed in Table 2.
. A perspective view of the repeating unit of the nickel polymer 2 is depicted in Fig. 5; selected structural parameters such as bond distances and angles for 1–3 are listed in Table 1. The various types of hydrogen bonds present in these coordination polymeric networks are listed in Table 2.
 | ||
| Fig. 5 Thermal ellipsoid plot of the repeating unit of the coordination polymer 2 with atom labelling scheme; polymers 1 and 3 are isostructural to 2. | ||
| M = Co; 1 | M = Ni; 2 | M = Zn; 3 | |
|---|---|---|---|
| M1–O2 | 2.087(2) | 2.0522(15) | 2.0733(19) |
| M1–O1 | 2.100(2) | 2.0707(16) | 2.106(2) |
| M1–O3 | 2.154(2) | 2.1091(14) | 2.1751(16) |
| O1′–M1–O1 | 180.0(1) | 180.0(1) | 180.0(1) |
| O2–M1–O2′ | 180.0(1) | 180.0(1) | 180.0(1) |
| O3–M1–O3′ | 180.0(1) | 180.0(1) | 180.0(1) |
| O2–M1–O1′ | 89.42(9) | 89.17(7) | 89.13(9) |
| O2–M1–O1 | 90.58(9) | 90.83(7) | 90.87(9) |
| O2–M1–O3 | 92.85(8) | 92.05(6) | 92.18(7) |
| O2′–M1–O3 | 87.15(8) | 87.95(6) | 87.82(7) |
| O1′–M1–O3 | 85.74(9) | 85.10(6) | 85.37(7) |
| O1–M1–O3 | 94.26(9) | 94.90(6) | 94.23(7) |
| Connectivity | D⋯A/Å | ∠D–H–A/° |
|---|---|---|
| O7–H13⋯O2 | 3.129(4) | 144.3 |
| N1–H6⋯O3 | 2.917(3) | 170.5 |
| C6–H8⋯O8 | 3.272(5) | 142.2 |
| O1–H1⋯O5 | 2.758(2) | 165.5 |
| O2–H4⋯O6 | 2.614(2) | 172.2 |
| O1–H2⋯O8 | 3.044(7) | 156.0 |
| O2–H3⋯O5 | 2.731(2) | 174.6 |
| N1–H7⋯O7 | 2.762(3) | 163.8 |
| C11–H11⋯O6 | 3.398(4) | 145.1 |
| O7–H12⋯O4 | 2.770(3) | 153.6 |
The repeating unit consists of a central metal ion (Co2+, Ni2+ or Zn2+) having its axial positions occupied by carboxylate groups and equatorial positions by water molecules to give an octahedral environment (Fig. 5). Thus, the BTEC ligand acts as a bridge in a unidentate terminal binding mode linking adjacent metal ions through its para positioned carboxylate groups, resulting in the formation of a coordination polymer.31 The remaining two carboxylate groups on each BTEC participate in an N–H⋯O hydrogen bond with intercalated diprotonated piperazinium dications. Thus a layered-solid consisting of alternating anionic metal–organic coordination polymer chains and cationic amine layers is formed (Fig. 6).
 | ||
| Fig. 6 Unit cell packing diagram for 3 showing all possible hydrogen bonds. | ||
Adjacent layers stack over one another in a staggered fashion, and are held together by N–H⋯O interactions between the free carboxylate of the BTEC and piperazinium cations, thus yielding a rigid and stable extended network containing sites responsible for, after dehydration, micropores or cavities. These sites are occupied by water molecules (four per unit cell) (Fig. 7). An extensive array of O–H⋯O hydrogen bonds also exist between the water molecules in the cavities and the uncoordinated carboxylate group of the BTEC ligands and the coordinated water molecules (Table 2). Besides the strong N–H⋯O and O–H⋯O hydrogen bonds, weak C–H⋯O hydrogen bonds32 are also present between the included water molecules and the carbon atoms on the piperazinium dications. Apart from the N–H⋯O interactions holding the metal–organic and cationic amine layers, numerous O–H⋯O hydrogen bonding interactions involving the coordinated water molecules of one M–BTEC layer and the carboxylate group of the adjacent M–BTEC layer lead to the formation of a tightly held metal–organic framework (Fig. 8). While the distance between two adjacent metal–organic polymer layers is 7.50 Å, the distance between the metal–organic and organic layers is 3.75 Å.
 | ||
| Fig. 7 Extended packing diagram of 2 (down z) showing the layered structure as well as the occluded water molecules. Several hydrogen bonds are omitted for clarity. | ||
 | ||
| Fig. 8 Three-dimensional packing of 1 in the crystal showing all the possible hydrogen bonds (down z). | ||
The bond distances found in these complexes 1–3 do not deviate appreciably from the expected values found in simple carboxylate complexes for these metal ions. For example, while the M–O bond lengths do not show appreciable deviation from those observed for previously synthesized BTEC complexes of Co2+ and Ni2+ ions, the O–M–O bond angles show appreciable deviation.25a,b The cis O–M–O angles around the metal atom in 2 vary from 89.17 to 94.90° with an average value of 90°. Other relevant metric parameters are listed in Table 1.
Conclusion
It has been shown in this contribution that a new family of coordination polymers can be readily synthesized under mild conditions from 1,2,4,5-benzenetetracarboxylic acid and divalent late transition metal salts (Co2+, Ni2+ and Zn2+) in the presence of a donor amine without employing hydrothermal synthesis or high pressures. The polymers have a layered structure consisting of alternating metal–ligand anionic and cationic amine layers. Although there are several neutral and cationic coordination polymers described in the literature, compounds 1–3 represent rare examples of anionic metal coordination polymeric networks. Heat treatment of the parent polymers results in the partial reversible loss of the water molecules. While dehydrated samples do not adsorb neutral gas molecules (e.g. CH4) and do not show a Type I isotherm, exposure of the dehydrated sample to liquid ammonia leads to the incorporation of ammonia molecules with the collapse of the framework structure. Further work in our laboratory is centred on the use of other polyamines as auxiliary ligands in the synthesis of newer coordination polymers apart from systematically investigating the role of auxiliary ligands in the formation of the resultant architecture of the polymeric networks.Experimental
General procedure
Water used as solvent in all the reactions was double distilled prior to use. Starting materials cobalt chloride hexahydrate (BDH), nickel chloride hexahydrate (Merck), zinc sulfate heptahydrate (Sarabhai Chemicals), piperazine hexahydrate (Koch-Light Laboratories), and pyromellitic anhydride (Fluka) were procured from commercial sources and used as received. All the starting materials and the reaction products described are air-stable and hence were routinely prepared under normal laboratory conditions without any precautions to exclude moisture or oxygen.Elemental analyses were performed on a Carlo Erba (Italy) Model 1106 Elemental Analyser. The melting points were measured in glass capillaries and are uncorrected values. Diffuse Reflectance UV-visible (DR UV) spectra were obtained on a Shimadzu UV 260 spectrophotometer. Infrared spectra were recorded on a Nicolet Impact 400 spectrometer as KBr diluted thin plates. Thermogravimetric analyses and differential thermal analyses were carried out at the RSIC, IIT-Bombay on a DuPont thermal analyser Model 2100 under a stream of nitrogen gas and a heating rate of 10 °C min−1. Gas adsorption studies were carried out at the Kyoto University using previously described methods.3
Syntheses
The nickel and zinc derivatives 2 and 3, respectively, were essentially synthesized following the same procedure as described for 1.
Dehydration and ammonia insertion experiments
Analytically pure samples of 1 and 2 were heated at approximately 60 °C (0.1 mmHg, 12 h). A large excess of ammonia gas was condensed into the flask containing the dehydrated sample at −50 °C. The resultant mixture was kept aside for 60 min with occasional stirring and the excess ammonia was boiled off.The same experiment can also be carried out just by exposing the dehydrated samples to gaseous NH3 to produce the same results as described above.
X-Ray structure analysis
Intensity data were collected on a Siemens-Stoe AED2 four-circle diffractometer using Mo-Kα radiation at 213 K for 1, and on Nonius MACH-3 diffractometer at room temperature (293 K) for compounds 2 and 3. The structure solution and refinement were carried out using SHELXS-9633 and SHELXL-97.34 All hydrogen atoms were identified from difference maps and were included in the successive refinement cycles. All non-hydrogen atoms (including those of solvent water molecules) were refined anisotropically, while all hydrogen atoms were assigned a uniform isotropic thermal parameter of 0.08. Other details pertaining to data collection, structure solution, and refinement are given in Table 3.CCDC reference numbers 169670–169672.
See http://www.rsc.org/suppdata/dt/b1/b105687p/ for crystallographic data in CIF or other electronic format.
| 1 | 2 | 3 | |
|---|---|---|---|
| a R1 = Σ|Fo| − |Fc|/Σ|Fo|; wR2 = [Σw(Fo2 − Fc2)/Σ(Fo2)2|1/2. | |||
| Empirical formula | C14H30CoN2O16 | C14H30N2NiO16 | C14H30N2O16Zn |
| Formula weight | 541.33 | 541.11 | 547.77 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group |
P |
P |
P |
| a/Å | 6.867(1) | 6.8436(4) | 6.847(5) |
| b/Å | 9.114(2) | 9.1166(7) | 9.122(1) |
| c/Å | 9.698(2) | 9.7167(6) | 9.769(2) |
| α/° | 104.63(3) | 105.346(6) | 104.87(1) |
| β/° | 98.04(3) | 97.746(5) | 98,16(3) |
| γ/° | 109.66(3) | 109.346(7) | 109.45(3) |
| V/Å3; Z | 535.9(2); 1 | 534.86(6); 1 | 538.5(4); 1 |
| T/K | 213(2) | 293(2) | 293(2) |
| μ/mm−1 | 0.886 | 0.993 | 1.225 |
| Total reflections | 3685 | 2059 | 2067 |
| Unique reflections | 1884 (Rint = 0.032) | 1882 (Rint = 0.004) | 1889 (Rint = 0.008) |
| R1, wR2 [I > 2σ(I)]a | 0.0437; 0.1119 | 0.0314; 0.0832 | 0.0343; 0.0921 |
| R1, wR2 (all data)a | 0.0438; 0.1121 | 0.0317; 0.0835 | 0.0346; 0.0924 |
Acknowledgements
This work was funded by the Council of Scientific and Industrial Research, New Delhi and Department of Science and Technology, New Delhi. R. M. thanks DAE for a Young Scientist Research Award. The authors also thank the Single Crystal X-ray Diffractometer Facility at IIT-Bombay for the intensity data of 2 and 3, and Professor S. Kitagawa for methane gas adsorption studies. Thanks are also due to Dr M. G. Walawalkar for useful input and suggestions at various stages of this work.References
- (a) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474 CrossRef CAS; (b) M. O'Keeffe, M. Eddaoudi, H. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3 CrossRef CAS and other articles in this issue; (c) D. Braga, J. Chem. Soc., Dalton Trans., 2000, 3705 RSC and other articles in this issue; (d) C. Janiak, Angew. Chem., Int. Ed. Engl., 1997, 36, 1431 CrossRef CAS; (e) P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638 CrossRef.
- (a) R. Murugavel, V. V. Karambelkar, G. Anantharaman and M. G. Walawalkar, Inorg. Chem., 2000, 39, 1381 CrossRef CAS; (b) R. Murugavel, V. V. Karambelkar and G. Anantharaman, Indian J. Chem. Sect. A: Inorg., Phys., Theor. Anal., 2000, 39, 843 Search PubMed; (c) R. Murugavel, G. Anantharaman, D. Krishnamurthy, M. Sathiyendiran and M. G. Walawalkar, Proc.-Indian Acad. Sci., Chem. Sci., 2000, 112, 273 Search PubMed; (d) R. Murugavel, K. Baheti and G. Anantharaman, Inorg. Chem., 2001, 40 , in press.
- (a) S.-I. Noro, S. Kitagawa, M. Kondo and K. Seki, Angew. Chem., Int. Ed., 2000, 39, 2081 CrossRef; (b) T. Okubo, S. Kitagawa, M. Kondo, H. Matsuzaka and T. Ishii, Angew. Chem., Int. Ed., 1999, 38, 931–933 CrossRef CAS; (c) M. Kondo, T. Okubo, A. Asami, S.-I. Noro, T. Yoshitomi, S. Kitagawa, T. Ishii, H. Matsuzaka and K. Seki, Angew. Chem., Int. Ed., 1999, 38, 140 CrossRef CAS; (d) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, O. M'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS.
- M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725 CrossRef CAS.
- C. V. K. Sharma and R. D. RogersChem. Commun., 1999, 83 Search PubMed.
- (a) G. B. Gardner, D. Venkataraman, J. S. Moore and S. Lee, Nature, 1995, 374, 792 CrossRef CAS; (b) D. Venkataraman, G. B. Gardner, S. Lee and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 11600 CrossRef CAS.
- (a) O. M. Yaghi, D. Richardson, T. L. Groy and Z. Sun, J. Am. Chem. Soc., 1994, 116, 807 CrossRef CAS; (b) O. M. Yaghi, R. Jernigan, H. Li, C. Davis and T. L. Groy, J. Chem. Soc, Dalton Trans., 1997, 2383 RSC; (c) M. Eddaoudi, H. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391 CrossRef CAS.
- D. M. L. Goodgame, D. A. Grachvogel and D. J. Williams, Angew. Chem., 1999, 111, 217 CrossRef; D. M. L. Goodgame, D. A. Grachvogel and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 153 CrossRef CAS.
- (a) S. R. Batten, B. F. Hoskins and R. Robson, Chem. Eur. J., 2000, 6, 156 CrossRef CAS; (b) S. R. Batten, B. F. Hoskins and R. Robson, Angew. Chem., 1997, 109, 652; S. R. Batten, B. F. Hoskins and R. Robson, Angew. Chem., Int. Ed. Engl., 1997, 36, 636 CrossRef CAS; (c) B. F. Hoskins, R. Robson and D. A. Slizys, J. Am. Chem. Soc., 1997, 119, 2952 CrossRef CAS.
- H. M. Colquhoun, R. A. Fairman, P. Tootell and D. J. Williams, J. Chem. Soc., Dalton Trans., 1999, 2651 RSC.
- (a) O. R. Evans, Z. Wang, R. Xiong, B. M. Foxman and W. Lin, Inorg. Chem., 1999, 38, 2969 CrossRef CAS; (b) O. R. Evans, Z. Wang and W. Lin, Chem. Commun., 1999, 1903 RSC; (c) M. A. Withersby, A. J. Blake, N. R. Champness, P. A. Cooke, P. Hubberstey, W. Li and M. Schroder, Inorg. Chem., 1999, 38, 2259 CrossRef CAS.
- L. R. MacGillivray, S. Subramanian and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1994, 1325 RSC.
- P. Brunet, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1997, 119, 2737 CrossRef CAS.
- Q. Wang, X. Wu, W. Zhang, T. Sheng, P. Lin and J. Li, Inorg. Chem., 1999, 38, 2223 CrossRef.
- P. Lightfoot and A. Snedden, J. Chem. Soc., Dalton Trans., 1999, 3549 RSC.
- M. J. Plater, M. St. J. Foreman, E. Coronado, C. J. Gomez-Garcia and A. M. Z. Slawin, J. Chem. Soc., Dalton Trans., 1999, 4209 RSC.
- J. Y. Lu, B. R. Cabrera, R. Wang and J. Li, Inorg. Chem., 1999, 38, 4608 CrossRef CAS.
- (a) H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571 CrossRef CAS; (b) T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651 CrossRef CAS; (c) R. H. Groeneman, L. R. MacGillivray and J. L. Atwood, Chem. Commun., 1998, 2735 RSC; (d) R. H. Groeneman, L. R. MacGillivray and J. L. Atwood, Inorg. Chem., 1999, 38, 208 CrossRef CAS.
- (a) O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096 CrossRef CAS; (b) O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703 CrossRef CAS; (c) O. M. Yaghi, C. E. Davis, G. Li and H. Li, J. Am. Chem. Soc., 1997, 119, 2861 CrossRef CAS; (d) C. J. Kepert and M. J. Rosseinsky, Chem. Commun., 1998, 31 RSC.
- M. J. Platers, R. A. Howie and A. J. Roberts, Chem. Commun., 1997, 893 RSC.
- (a) H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 1998, 120, 10622 CrossRef CAS; (b) H. J. Choi, T. S. Lee and M. P. Suh, Angew. Chem., Int. Ed., 1999, 38, 1405 CrossRef CAS.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1996, 118, 295 CrossRef CAS.
- (a) L. Deakin, A. M. Arif and J. S. Miller, Inorg. Chem., 1999, 38, 5072 CrossRef CAS; (b) M. R. St. J. Foreman, T. Gelbrich, M. B. Hursthouse and M. John Plater, Inorg. Chem. Commun., 2000, 3, 234 CrossRef.
- (a) H. Li, C. E. Davis, T. L. Groy, D. G. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 2186 CrossRef CAS; (b) B. Chen, M. Eddaoudi, T. M. Reineke, J. W. Kampf, M. O. Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 11559 CrossRef CAS.
- (a) C. Robl and S. Hentschel, Mater. Res. Bull., 1991, 26, 1355 CrossRef CAS; (b) D. Poleti and L. Karanovic, Acta Crystallogr., Sect. C, 1989, 45, 1716 CrossRef; (c) D. L. Ward and D. C. Leuhrs, Acta Crystallogr., Sect. C., 1983, 39, 1370 CrossRef; (d) C. Robl, Mater. Res. Bull., 1992, 22, 99 CrossRef.
- (a) D. Poleti, D. R. Stojakovic, B. V. Prelesnik and R. M. Herak, Acta Crystallogr., Sect. C., 1988, 44, 242 CrossRef; (b) S. M. Jessen and H. Kuppers, Acta Crystallogr., Sect. C, 1990, 46, 2351 CrossRef; (c) D. Chu, J. Xu, L. Duan, T. Wang, A. Tang and L. Ye, Eur. J. Inorg. Chem., 2001, 1135 CrossRef CAS.
- (a) F. D. Rochon and G. Massarweh, Inorg. Chim. Acta, 2001, 314, 163 CrossRef CAS; (b) M. J. Plater, M. R. St. J Foreman, R. A. Howie, J. M. S. Skakle and A. M. Z. Slawin, Inorg. Chim. Acta, 2001, 315, 126 CrossRef CAS; (c) B. T. Usubaliev, A. N. Shnulin and H. S. Mamadow, Koord. Khim., 1982, 8, 1532 Search PubMed; (d) C. Robl, Z. Anorg. Allg. Chem., 1987, 554, 79 CrossRef CAS; (e) J. Z. Zou, Q. Liu, Z. Xu, X. Z. You and X. Y. Huang, Polyhedron, 1998, 17, 1863 CrossRef CAS; (f) S. M. Jessen, H. Kuppers and D. C. Luehrs, Z. Naturforsch., Teil B, 1992, 47, 1141 Search PubMed; (g) L. Karanovic, D. Poleti, G. A. Bogdanovic and A. Spasojevic-de Bire, Acta Crystallogr., Sect. C, 1999, 55, 911 CrossRef; (h) W. Ge-Cheng, J. Zhong, D. Zhi-Bang, Y. Kui-Yue and N. Jia-Zan, Jiegou Huaxue, 1991, 10, 106 Search PubMed; (i) P. Chaudhuri, K. Oder, K. Wieghardt, S. Gehring, W. Haase, B. Nuber and J. Weiss, J. Am. Chem. Soc., 1988, 110, 3657 CrossRef CAS; (j) F. Jaber, F. Charbonnier and R. Faure, J. Chem. Crystallogr., 1997, 27, 397 CAS; (k) W. Chen, N. Heng Tioh, J.-Z. Zou, Z. Xu and X.-Z. You, Acta Crystallogr., Sect. C, 1996, 52, 43 CrossRef.
- D. C. Luehrs and C. S. Day, Inorg. Chim. Acta, 1988, 142.
- (a) M. Matzapetakis, C. P. Raptopoulou, A. Terzis, A. Lakatos, T. Kiss and A. Salifoglou, Inorg. Chem., 1999, 38, 618 CrossRef CAS; (b) C. Djordjevic, M. Lee and E. Sinn, Inorg. Chem., 1989, 28, 719 CrossRef CAS; (c) G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 2nd Edn., 1984 Search PubMed.
- The BTEC ligand behaves more like terephthalic acid by coordinating to the metal through only two of its carboxylic acid groups. For examples of coordination polymers formed by terephthalic acid, see ref. 18.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS.
- G. M. Sheldrick, SHELXS-96: Program for Crystal Structure Solution, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXL-97: Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |