Intramolecular sensitisation of lanthanide(III) luminescence by acetophenone-containing ligands: the critical effect of para-substituents and solvent
Andrew
Beeby
*,
Lisa M.
Bushby
,
Davide
Maffeo
and
J. A.
Gareth Williams
*
Department of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE
First published on 28th November 2001
Abstract
Tetraazamacrocyclic ligands have been prepared in which three of the four nitrogen atoms are functionalised with carboxylate donors and the fourth is alkylated with a para-substituted acetophenone group {–CH2C(O)C6H4–X, where X = H, OMe, NMe2}. The europium(III), gadolinium(III) and (for X = H) terbium(III) complexes of these potentially octadentate ligands have been prepared. The unsubstituted and methoxy-substituted ligands sensitise the europium(III) emissive state with high efficiency in aqueous solution. Calculation of the pure radiative lifetime, based on the contribution of the 5D0
→
7F1 transition to the total integrated emission intensity, has allowed an estimate of the efficiency of energy transfer to be made. Sensitisation of europium luminescence also occurs in the complex of the dimethylamino-substituted
ligand but the process is highly solvent-dependent: very efficient sensitisation is observed in dichloromethane and DMSO, but the complex is non-emissive in water and only slightly emissive in acetonitrile. UV-Visible absorption spectra indicate that in all the solvents investigated, the carbonyl oxygen of the ligand is directly coordinated to the metal ion. The intensity of the hypersensitive 5D0
→
7F2 transition in the three complexes mirrors the degree of polarisation of the ketone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.
O bond.
Introduction
Although first observed 60 years ago,1 the sensitisation of lanthanide luminescence by organic chromophores is a subject which continues to attract a great deal of attention, owing primarily to its potential use in luminescent probes and sensors and for light amplification and frequency conversion.2–4 Two characteristic properties of lanthanide luminescence are central to such applications, namely the long lifetimes of emission under ambient conditions and the line-like nature of the emission spectra. Many applications require water-soluble complexes and this poses a challenge in the design and preparation of suitable complexes, since the lanthanide(III) ions have a high affinity for water molecules as ligands and their excited states are efficiently quenched by water. Successful complexes will have high thermodynamic and kinetic stability with respect to metal ion dissociation and ideally saturate the coordination shell of the metal ion, to prevent water molecules from binding. The incorporation of appropriate organic chromophores into the complex to act as efficient sensitisers of the lanthanide excited states is also an important objective in overcoming the problem of the very low extinction coefficients of the Ln3+ ions (typically ε < 1).Studies on several chromophores have demonstrated the importance of the triplet state in the energy transfer process.5 The overall quantum yield of luminescence, ϕlum, is determined by the triplet yield of the chromophore (ϕT), the efficiency of energy transfer (ηET) and the efficiency of metal centred luminescence (ηLn):
| φ lum = φT·ηET·ηLn | (1) |
We have recently demonstrated that aromatic ketones with n,π* triplet states, which have high triplet quantum yields, are particularly suitable as sensitisers,6 provided that there is a reasonable match between the triplet energy and the emissive level (or a higher excited level) of the lanthanide such that ηET is also optimal (the energy gap should, however, be at least 1500 cm−1 if back energy transfer to the chromophore is to be minimised). A striking recent example of ketone-sensitised lanthanide luminescence is to be found in the 1 ∶ 1 complex formed between Eu(fod)3 (Hfod = 1,1,1,2,2,3,3-heptafluoro-7,7-dimethyloctane-4,6-dione) and Michler's ketone, 4,4′-bis(N,N′-dimethylamino)benzophenone, in benzene solution.7 Here, the substituted benzophenone acts as an auxiliary ligand and sensitises the metal ion very efficiently. Our recent work investigated stable, water soluble Eu3+ and Tb3+ complexes of a ligand incorporating a benzophenone group covalently-bound via an amide linker, which gave impressive quantum yields in aqueous solution.6 We report here on the preparation of related tetraazamacrocyclic ligands incorporating para-substituted acetophenone groups directly linked (via the CH2 group) to one of the nitrogen atoms of the macrocycle, such that the ketone oxygen can potentially act as a donor to a bound lanthanide ion. The ketone-sensitised luminescence of the europium complexes of these ligands is described.
Results and discussion
Synthetic routes
Ligands based on the tetraazamacrocycle 1,4,7,10-tetraazacyclododecane (cyclen) tri- or tetra-N-substituted with carboxylate donors are well-known to form lanthanide(III) complexes of exceptional kinetic and thermodynamic stability.3,8,9 For example, the Gd3+ complex of the tetraacetate is sufficiently stable for use in vivo in millimolar quantities as a contrast agent for MRI.9 The ketone-substituted ligands L1–L3 were synthesised in order to explore the efficacy of acetophenones as sensitisers of the metal excited state in the europium complexes. The synthetic routes to the compounds are summarised in Scheme 1. Reaction of cyclen with three equivalents of tert-butylbromoacetate in chloroform at room temperature for 72 hours, in the presence of one equivalent of potassium carbonate, led primarily to the tert-butyl ester of DO3A, 4, which could be separated from small amounts of the tetra- and di-alkylated macrocycle by chromatography on silica. The ester was subsequently alkylated with the appropriate α-bromoacetophenone, 1a–3a, in acetonitrile in the presence of a catalytic amount of KI and potassium carbonate as the base. Compounds 2a and 3a were prepared from the parent para-substituted acetophenones. It is well-known that the bromination of arylmethylketones often leads to a complex mixture of products because aromatic ring substitution and bromine addition to the enol form often compete effectively with radical side-chain halogenation. Competitive ring bromination is particularly facile in amino-substituted compounds, due to the activating effect of the electron-donating amino group. We adopted routes to these compounds based on recently described procedures for the efficient, selective preparation of side-chain brominated compounds. The para-methoxy-substituted compound was prepared by bromination of 4′-methoxyacetophenone using HBr in the presence of tert-butylhydroperoxide, a combination of reagents that has been reported recently for in situ generation of positive halogen species avoiding the hazards associated with the preparation of tert-butylhypobromite, and which leads to selective side-chain bromination of acetophenone itself, with no ring bromination.10 We found this method to be equally selective for side-chain bromination of the para-methoxy substituted compound. 2-Bromo-4′-dimethylaminoacetophenone was prepared in three steps from 4′-dimethylaminoacetophenone according to a literature procedure.11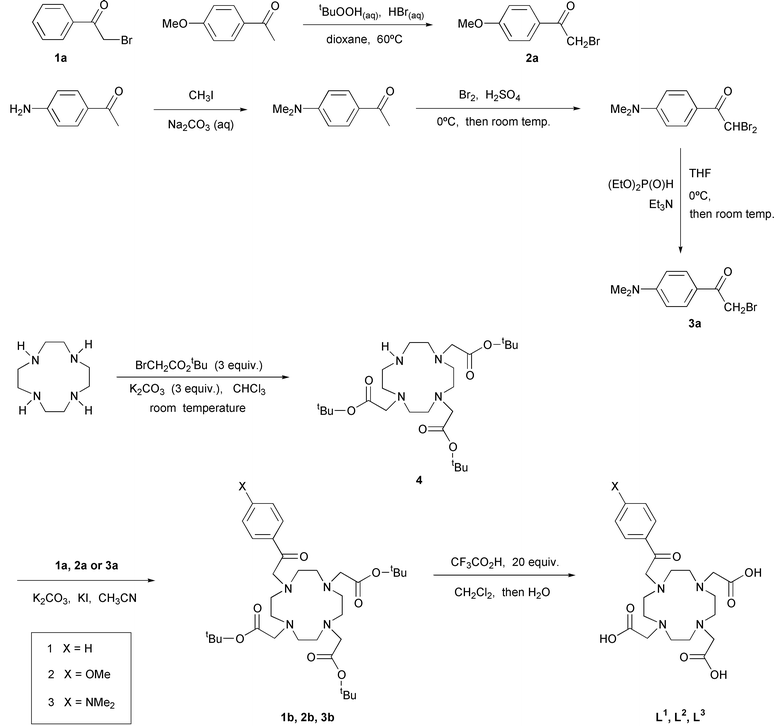 | ||
| Scheme 1 | ||
Following alkylation of the secondary amine in 4 with the α-bromoketones, the tert-butyl esters (1b–3b) were cleaved using trifluoroacetic acid, to generate the ligands L1–L3. The europium and gadolinium complexes were prepared by reaction with an equimolar amount of the lanthanide(III) nitrate in aqueous solution and subsequently purified by chromatography on a short column of alumina. The terbium(III) complex of L1 was prepared similarly. All the complexes were characterised by electrospray mass spectrometry. Although the large paramagnetism and severe line-broadening associated with the gadolinium(III) and terbium(III) ions prohibited NMR analysis, the smaller shifts and narrower line-widths induced by europium(III) allowed the 1H NMR spectra of the europium complexes to be recorded, revealing in each case the well-established pattern of resonances characteristic of square-antiprismatic complexes found with related ligands.12
Photophysical properties
The more strongly donating dimethylamino group has a much larger effect on the π–π* singlet excited state in L3, shifting the band to 339 nm. Moreover, the band is more red-shifted in the complexes by 30 nm, with a maximum at 372 nm and extending to beyond 400 nm (Fig. 1). This is indicative of a much stronger interaction of the carbonyl group with the metal ion in this case, giving rise to a complex with a higher charge-transfer character. For example, an effect of this type has been reported recently upon coordination of Michler's ketone [4,4′-bis(N,N-dimethylamino)benzophenone] to Eu(fod)3 in benzene, where a ground-state complex is formed characterised by an intense absorption band centred at 414 nm.7 The authors attribute this to a bathochromic shift of the first singlet–singlet excited state noting that, since excitation of this transition moves the electron density towards the carbonyl group, the presence of the lanthanide ion would be expected to have a large effect on its energy. However, coordination only occurred in non-coordinating solvents. In contrast, in the present instance, the carbonyl group remains bound even in aqueous solution, presumably because the seven other ligating sites of the macrocyclic ligand are able to maintain it in a position suitable for binding.
 | ||
Fig. 1 Absorbance spectra of L3 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and of EuL3 (—) in ethanol solution at 293 K. ) and of EuL3 (—) in ethanol solution at 293 K. | ||
Such geometric factors should also favour binding of the carbonyl group in the complexes of L1 and L2, despite the fact that no shifts in the absorption bands are observed in these cases. The absence of a red-shift in these complexes does not, however, mean that the carbonyl group remains uncoordinated, as evident from the following control experiment. Upon mixing 4,4′-dimethoxybenzophenone and Eu(fod)3 in toluene, no significant red-shifts nor new bands were observed in the absorption spectra, in contrast to the observations with Michler's ketone. Nevertheless, the europium emission intensity was substantially increased upon excitation at 365 nm, indicative of sensitisation of the metal by the benzophenone and hence complex formation by coordination of the ketone to the lanthanide ion. Moreover, the two complexes were found to contain only one metal-coordinated water molecule (vide infra), in contrast to the two water molecules found in complexes of related ligands offering heptadentate coordination (e.g. those of 1,4,7,10-tetraazacyclododecane-1,4,7-triacetate, DO3A), lending further support to the proposal that the ketone oxygen is indeed coordinated to the metal.
Lifetimes of emission are summarised in Table 1. From the values in H2O and D2O, the hydration number q (which provides an indication of the number of metal-bound water molecules) was determined using the Horrocks equation13 to be 1.24 and 1.20 for EuL1 and EuL2 respectively. After correction for the weaker effect of outer-sphere water molecules,14 these values are reduced to 1.10 and 1.05 respectively, clearly indicating that there is one metal-bound water molecule in both cases. This is consistent with observations on the complexes of structurally related octadentate amide ligands where a metal-bound water molecule increases the coordination number to nine12 and, as noted above, supports the proposal that the carbonyl oxygen is coordinated to the metal, preventing the entry of a second water molecule.
| Complex | E T/cm−1b | Absorbance λmax/nm | τ H2O/msc | τ D2O/msc | q d | ϕ H2O e | ϕ D2O e |
|---|---|---|---|---|---|---|---|
| a For the terbium complex of ligand L1: τH2O = 1.60 ms, τD2O = 2.6 ms, q = 0.98; ϕH2O = 0.12, ϕD2O = 0.23. b Triplet energy of the acetophenone chromophore ±200 cm−1, as estimated from the phosphorescence spectra of the gadolinium complexes at 77 K. c Lifetimes of the europium-centred emission monitored at 593 nm, 293 K; uncertainty ±5%. d q is the hydration state (number of coordinated water molecules) as determined from τH2O and τD2O using the equation of Parker et al.14 e Quantum yields of the europium complexes at 293 K, measured using cresyl violet in methanol and rhodamine 101 in acidified ethanol as standards; uncertainty ±10%. f EuL3 was non-emissive in aqueous solution. | |||||||
| LnL1 | 25600 | 265 | 0.62 | 2.26 | 1.10 | 0.006 | 0.021 |
| 25300 | |||||||
| LnL2 | 24500 | 305 | 0.63 | 2.18 | 1.05 | 0.098 | 0.34 |
| 24500 | |||||||
| LnL3 | 21000 | 375 | f | — | — | — | — |
| 20000 | |||||||
The emission spectra of the two complexes EuL1 and EuL2 were very similar (Fig. 2). A sharp 5D0 → 7F0 band of modest intensity at 579.8 nm is observed in each case. The relative contribution of the purely magnetic dipole 5D0 → 7F1 transition to the total integrated emission intensity is similar for both complexes, but the hypersensitive 5D0 → 7F2 band is rather more intense in EuL2 (vide infra).
 | ||
| Fig. 2 Emission spectra of EuL1 (a) and EuL2 (b) in solution in D2O at 293 K. The excitation wavelength was 265 and 305 nm respectively; excitation and emission bandpasses of 2.5 nm. | ||
Overall quantum yields of metal-centred luminescence, ϕlum, upon excitation of the acetophenone group are given in Table 1. In order to assess the efficiency of energy transfer ηET using these quantum yields, an estimate of both ϕT and ηLn is required [eqn. (1) above]. Given that the sensitiser has an n,π* triplet state, and also that the lanthanide ions have high spin–orbit coupling constants, it is appropriate to assume that ϕT (the quantum yield of triplet formation) is close to 1.6 The efficiency of metal-centred luminescence, ηLn, can be calculated from the observed emission lifetime τobs, provided that the pure radiative lifetime τR is available, since:
| ηLn = τobs/τR | (2) |
| kR = ΣJA (0,J) | (3) |
The relative contribution of each 5D0 → 7FJ transition to the spectrum, the branching ratio β, is the ratio of the intensity of the band to the total intensity in the corrected emission spectrum, and is determined by the spontaneous emission probability:
| β(0,J) = A(0,J)/ΣJA(0,J) = A(0,J)/kR = A(0,J)·τR | (4) |
The forbidden lanthanide(III) (f–f) emission bands consist of magnetic dipole (MD) and induced electric dipole (ED) transitions. The former are virtually independent of the ion's surroundings, in contrast to the latter where the geometry and ligating atoms have a significant effect on the transition probabilities. The 5D0 → 7F1 (J1) band of Eu3+ centred at 593 nm is entirely MD and consequently has an oscillator strength that is independent of the ligand field and complex symmetry.16 Thus, given a value for the spontaneous emission probability A(0,1) of the 5D0 → 7F1 transition and using eqn. (4), it is possible to estimate τR from the relative contribution of this band to the experimentally determined emission spectrum:
| 1/τR = A(0,1)/β(0,1) = A(0,1)[Itot/I(0,1)] | (5) |
For complexes EuL1 and EuL2, the results of these calculations [eqns. (1)–(3)], using the relative contribution of the J1 band in the corrected emission spectrum together with the experimentally determined values of τobs and ϕlum, leads to the values of τR, ηLn, ηET and Σknr given in Table 2. It can be seen that the energy transfer efficiencies are high in each case and especially so for the methoxy-substituted system. The introduction of the methoxy substituent lowers the triplet energy by about 1000 cm−1 (Table 1), leading to a better match with the acceptor 5D1 and 5D0 states of the europium ion. As expected, ηET does not change significantly on going from H2O to D2O, and the main factor that limits the observed luminescence quantum yield of these complexes is the rather low efficiency of metal-centred luminescence, even in D2O.
| EuL1 | EuL2 | |||
|---|---|---|---|---|
| H2O | D2O | H2O | D2O | |
| a [I(0,1)/Itot] is the relative contribution of the 5D0 → 7F1 band intensity, I(0,1), to the total integrated emission intensity, Itot; τR is the pure radiative lifetime determined from [I(0,1)/Itot] using the method described in the text; kR is the radiative rate constant = 1/τR; ηLn is the efficiency of metal-centred luminescence obtained using eqn. (2); ηET is the efficiency of energy transfer calculated using eqn. (1) and assuming that ϕT = 1; Σknr (the sum of the non-radiative decay constants) = [(1/τobs) − kR]. | ||||
| [I(0,1)/Itot] | 0.24 | 0.25 | 0.22 | 0.21 |
| τ R/ms | 7.37 | 7.74 | 6.65 | 6.62 |
| k R/s−1 | 136 | 129 | 150 | 151 |
| τ obs/ms | 0.62 | 2.26 | 0.63 | 2.18 |
| η Ln | 0.084 | 0.29 | 0.094 | 0.33 |
| ϕ lum | 0.058 | 0.21 | 0.093 | 0.34 |
| η ET | 0.71 | 0.72 | 0.99 | 1.0 |
| Σknr/s−1 | 1480 | 313 | 1440 | 308 |
C–O− character of the ketone carbonyl group upon introduction of the increasingly electron-donating methoxy and dimethylamino substituents. The observed trend confirms, then, that the ketone group is coordinated to the metal ion in each
of the three complexes.
| EuL1 | EuL2 | EuL3 | |
|---|---|---|---|
| J0/J1 | 1.11 | 0.40 | 0.15 |
| J2/J1 | 0.67 | 1.43 | 3.22 |
| J3/J1 | 0.20 | 0.23 | 0.23 |
| J4/J1 | 0.78 | 0.97 | 2.74 |
 | ||
| Fig. 3 Emission spectrum of EuL3 in solution in ethanol at 293 K; λex = 370 nm; bandpass = 2.5 nm. | ||
The total emission intensity of EuL3 was found to be very sensitive to the solvent. In particular, despite the reasonable quantum yields in non-aqueous solvents (Table 4), no emission was detectable in aqueous solution (H2O or D2O). For the other solvents examined, luminescence was observed in each case, although the intensity varied erratically (Fig. 4), showing no correlation with the dielectric constant, nor with other solvent parameters such as that of Reichardt. Monoexponential decay of the emission was observed in DMSO and alcohol solutions, and the measured lifetime was found to vary substantially with solvent (Table 4). Emission in CH3CN, CH2Cl2 and CHCl3 displayed non-exponential decay kinetics, possibly due to the slow exchange between the two species with and without a metal-bound water molecule.20
 | ||
| Fig. 4 Integrated emission intensity of EuL3 in several solvents (λex = 370 nm) plotted against dielectric constant. | ||
The very sensitive dependence of the emission intensity and lifetime on solvent is probably a combination of effects, as revealed by applying the above analysis of spectra and lifetimes to EuL3. The results of this treatment are summarised in Table 4, which show that the solvent has not only an effect on the non-radiative decay pathways, but also a profound influence on both the pure radiative lifetime and the efficiency of energy transfer. The pure radiative lifetime is significiantly shorter in DMSO than in alcohols, whilst the non-radiative decay pathways (Σknr) are less significant, which together account for the substantially higher quantum yield observed in DMSO. The efficiency of energy transfer is close to unity.
Conclusion
In summary, the complexes reported here represent a new class of compound in which an acetophenone group covalently linked to the macrocyclic ligand is able to bind to the coordinated lanthanide ion, promoting efficient energy transfer from chromophore to metal. Introduction of a methoxy substituent into the para position of the acetophenone further increases the efficiency, whilst a dimethylamino substituent leads to high emission intensities in non-aqueous solvents only.Experimental
General
Cyclen was supplied by Strem and used as supplied. The preparation of 4′-dimethylamino-2-bromoacetophenone followed procedures described recently in the literature starting from 4′-aminoacetophenone,11 which was obtained from Aldrich. 2-Bromoacetophenone and 4′-methoxyacetophenone are also available commercially. Chromatography was carried out on silica gel (60, 40–63 μm, Fluorochem) or on alumina (neutral, Brockmann I). Solvents were Analar or HPLC grade, and water was purified by the Milli Q system. Proton and 13C NMR spectra were recorded on a Varian Mercury-200 (200 and 50.3 MHz respectively) or a Unity-300 (300 and 75.5 MHz). Proton spectra were referenced to residual protio solvent resonances and 13C spectra to the solvent carbon resonance. Electrospray mass spectra were measured on a VG Platform II instrument. UV-Visible absorbance spectra were recorded using a Unicam UV-2 spectrometer using quartz cuvettes of 1 cm pathlength. Steady-state emission spectra were recorded using an Instruments S.A. Fluromax and lifetimes were measured using a Perkin-Elmer LS 50B instrument. An Oxford Instruments variable temperature liquid nitrogen cryostat (DN1704) was used for recording the phosphorescence spectra of the chromophores at 77 K. Elemental analyses were carried out using an Exeter Analytical E-440 Analyser.Syntheses
O), 164.2 (C–COCH2), 132.3 and 131.4 (aromatic CH), 114.1 (C–OMe), 55.7 (CH2), 30.9 (CH3). m/z (ES+): 253 (100%, M + Na+).
For example, for [EuL3], δH (D2O, 200 MHz): 36.3 (1 H), 32.0 (1 H), 31.4 (1 H), 30.6 (1 H) (Hax); 6.1 (4 H, aromatic H); 3.3 (6 H, N(CH3)2); 1.8 (1 H), −1.2 (1 H), −1.7 (1 H), −2.6 (1 H), −3.0 (1 H), −4.6 (1 H), −6.2 (1 H), −7.1 (1 H), −8.8 (3 H overlapping), −10.4 (1 H), −11.5 (1 H), −12.9 (1 H), −14.8 (4 H overlapping), −15.7 (1 H), −16.7 (1 H) (8 Heq, remaining 4 Hax, CH2CO). m/z (ES+): 680.1612 (M + Na+, calculated value for C24H34N5O7EuNa = 680.1568).
Similarly, m/z (ES+): EuL1: 635 (M + Na+); GdL1: 618 (M + H+); TbL1: 619 (M + H+); EuL2: 667.1266 (M + Na+, calculated value for C23H31O8N4EuNa = 667.1252); found C 36.82, H 4.89, N 7.32% (C23H31O8N4Eu·5H2O requires C 37.60, H 5.59, N 7.63%); GdL2: 671 (M + Na+); found C 36.84, H 4.82, N 7.45% (C23H31O8N4Gd·5H2O requires C 37.35, H 5.55, N 7.58%); GdL3: 685 (M + Na+).
Acknowledgements
We thank the EPSRC and Opsys Limited for a CASE award (L. M. B.), the University of Durham for a studentship (D. M.) and the Royal Society and the Nuffield Foundation for grants towards the purchase of equipment.References
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214 CrossRef CAS.
- N. Sabbatini, M. Guardigli and J.-M. Lehn, Coord. Chem. Rev., 1993, 123, 201 CrossRef CAS.
- D. Parker and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1996, 3613 RSC.
- M. P. Oude Wolbers, F. C. J. M. van Veggel, F. G. A. Peters, E. S. E. van Beelen, J. W. Hofstraat, F. A. J. Geurts and D. N. Reinhoudt, Chem. Eur. J., 1998, 4, 772 CrossRef CAS.
- For example: B. Alpha, R. Ballardini, V. Balzani, J.-M. Lehn, S. Perathoner and N. Sabbatini, Photochem. Photobiol., 1990, 52, 299 Search PubMed; A. Beeby, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1565 Search PubMed; A. Beeby, S. Faulkner, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 2001, 1268 RSC.
- A. Beeby, L. M. Bushby, D. Maffeo and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 2000, 1281 RSC.
- M. H. V. Werts, M. A. Duin, J. W. Hofstraat and J. W. Verhoeven, Chem. Commun., 1999, 799 RSC.
- For a review, see: V. Alexander, Chem. Rev., 1995, 95, 273 Search PubMed.
- Gadolinium(III) complexes for use in MRI, including those of DOTA and related ligands, have been the subject of a recent comprehensive review: P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 Search PubMed.
- N. B. Barhate, A. S. Gajare, R. D. Wakharkar and A. V. Bedekar, Tetrahedron, 1999, 55, 11127 CrossRef CAS.
- Z. Diwu, C. Beachdel and D. H. Klaubert, Tetrahedron Lett., 1998, 39, 4987 CrossRef CAS.
- R. S. Dickins, J. A. K. Howard, C. L. Maupin, J. M. Moloney, D. Parker, J. P. Riehl, G. Siligardi and J. A. G. Williams, Chem. Eur. J., 1999, 5, 1095–1105 CrossRef CAS.
- W. De W. Horrocks, Jr. and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- W. T. Carnall, in Handbook on the Physics and Chemistry of Rare Earths, eds. K. A. Gschneidner, Jr. and L. Eyring, North Holland Publishing Co., Amsterdam, 1979, ch. 24 Search PubMed; A similar treatment has been applied by others; for example, M. H. V. Werts, Ph.D. Thesis, University of Amsterdam, 2000 Search PubMed.
- (a) R. D. Peacock, Struct. Bonding (Berlin), 1975, 22, 83 Search PubMed; (b) A. F. Kirby, D. Foster and F. S. Richardson, Chem. Phys. Lett., 1983, 95, 507 CrossRef CAS.
- The methods by which the spontaneous emission probabilities may be calculated have been discussed widely, for example, ref. 15 or: M. J. Weber, T. E. Varitimos and B. H. Matsinger, Phys. Rev. B, 1973, 8, 47 Search PubMed.
- A. F. Kirby and F. S. Richardson, J. Phys. Chem., 1983, 87, 2544 CrossRef CAS.
- The dynamic coupling model has been developed in terms of a polarisability tensor α(L) of the ligand: S. F. Mason, R. D. Peacock and B. Stewart, Mol. Phys., 1975, 30, 1829 Search PubMed.
- A. S. Batsanov, A. Beeby, J. I. Bruce, J. A. K. Howard, A. M. Kenwright and D. Parker, Chem. Commun., 1999, 1011 RSC.
- M. Woods, Ph.D. Thesis, University of Durham, 1998 Search PubMed.
Footnotes |
| † EPA refers to the solvent system: ethanol/isopentane/diethyl ether in the ratio 2/5/5 by volume. This solvent offers excellent properties for measurement of spectra and lifetimes at low temperature, with little tendency for cracking. The solubility of the complexes in EPA, although minimal, is sufficient to obtain solutions with suitable absorbances. |
| ‡ Application of the Einstein coefficient to express the rate of relaxation from an excited state ψJ to a final state ψJ′ under a magnetic dipole mechanism leads to: A(ψJ, ψJ′) = {64π4σ3/3h(2J + 1)}·n3·Smd(ψJ, ψJ′), where σ is the energy gap between the two states (cm−1), n is the refractive index of the medium and Smd(ψJ, ψJ′) is the magnetic dipole line strength.17 This last quantity has been calculated theoretically for the 5D0 → 7F1 transition of Eu3+ in a number of studies and verified experimentally; we have used the value of Kirby and Richardson18 (884 × 10−8 Debye2) together with an energy of 17010 cm−1 as the baricentre of the transition and n = 1.334 for H2O, leading to the value of 32.4 s−1. |
| This journal is © The Royal Society of Chemistry 2002 |