Formation of 1 ∶ 1 and 2 ∶ 2 complexes of Ce(III) with the heteropolytungstate anion α2-[P2W17O61]10−, and their interaction with proline. The structure of [Ce2(P2W17O61)2(H2O)8]14−†
Masahiro Sadakane, Angelo Ostuni and Michael T. Pope*
Department of Chemistry, Box 571227, Georgetown University, Washington D.C. 20057-1227, USA. E-mail: popem@georgetown.edu; Tel: +1-202-687-6253; Fax: +1-202-687-6209
First published on 6th December 2001
Abstract
The “1 ∶ 1” complex of Ce3+ and α2-[P2W17O61]10− is formed in an equimolar solution of Ce(NO3)3 and K10[P2W17O61], but attempts to isolate a solid yield salts of the 1 ∶ 2 complexes first described by Peacock and Weakley in 1971. With a 3-fold excess of Ce3+ however, a crystalline ammonium salt (anion 1) could be isolated in 90% yield based on tungstate. Structural analysis of these crystals (monoclinic, C2/c; a
= 46.1124(6); b
= 12.7451(1); c
= 26.4624(2) Å, β
= 93.570(1)°, Z
= 4; 18856 independent reflections, R1 = 0.0799; wR2 = 0.1594) revealed a head-to-head dimeric
anion with each 9-coordinate Ce(III) cation attached to four oxygens that surround the lacuna of one tungstophosphate anion, a terminal O(W) of the second tungstophosphate, and four water molecules. Phosphorus-31 NMR spectra of solutions of 1 revealed two sets of resonances, A and B, the relative intensities of which could be fitted to an equilibrium 2A ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B, with K
= 1.46 ± 0.04 M−1 at 23 °C. Further NMR analysis suggests a scheme in which 1, which is in rapid exchange with the “monomer” (2), is also in slow exchange with a labile associate of Ce3+ and the 1 ∶ 2 complex. The exchange process between 1 and 2 is responsible for the non-observance of one of the expected
9 lines in the tungsten-183 NMR spectrum and an exchange-broadened P-NMR line. Addition of proline to form a ternary complex [Ce(P2W17O61)(proline)(aq)]7−(Kform
= 4.5 ± 0.1 M−1 from 1H-NMR) restores the missing W-NMR resonance.
B, with K
= 1.46 ± 0.04 M−1 at 23 °C. Further NMR analysis suggests a scheme in which 1, which is in rapid exchange with the “monomer” (2), is also in slow exchange with a labile associate of Ce3+ and the 1 ∶ 2 complex. The exchange process between 1 and 2 is responsible for the non-observance of one of the expected
9 lines in the tungsten-183 NMR spectrum and an exchange-broadened P-NMR line. Addition of proline to form a ternary complex [Ce(P2W17O61)(proline)(aq)]7−(Kform
= 4.5 ± 0.1 M−1 from 1H-NMR) restores the missing W-NMR resonance.
Introduction
The chemistry of polyoxometalates continues to attract interest as a result of their realized and potential applications in fields as disparate as catalysis, materials science, structural biology, and medicine.1 In recent years attention has been focussed on very large polyoxoanions which span the interface between discrete molecular species and extended lattices, and which can demonstrate novel host–guest chemistry, and unusual optical, magnetic and other properties.2 The structures of such large polyoxoanions can be mentally decomposed into smaller discrete “building blocks” with various linking atoms or groups. Simple, early examples of such composite polyoxometalates are complexes such as [Mn(Nb6O19)2]12−3 and [Eu(PW11O39)2]11−,4 in which the linkers are transition metal and lanthanide cations. The latter type of complexes, first reported by Peacock and Weakley, are easily synthesized from the lacunary polyoxoanion “ligand” and the lanthanide cation which adopts a square prismatic 8-coordinate coordination geometry. Large numbers of such complexes with lanthanide and actinide(4+) cations and a variety of polyoxometalate ligands have been characterized since the first reports.2,5 Much research has been directed at the use of the tungstophosphate anion [P2W17O61]10− (α2 isomer) for radioactive waste treatment via sequestration of transuranium elements.6Although Peacock and Weakley had noted the existence of 1 ∶ 1 complexes such as [Ce(PW11O39)(H2O)x]7−, and these were later characterized by spectroscopic and electrochemical methods,7,8 the first structural studies revealed that the anions assembled into linear polymeric arrays upon crystallization.9 We have recently examined the 1 ∶ 1 complex of the metastable α1 isomer of [P2W17O61]10− with Ce3+ and showed that it crystallizes in the form of a head-to-head dimer, [{Ce(H2O)4(P2W17O61)}2]14−.10 Very recently, Francesconi et al. have described the structure of [{Eu(H2O)3(P2W17O61)}2]14−.11
This paper describes the synthesis, and solution and solid state characterization of the 1 ∶ 1 complex of the α2 isomer of [P2W17O61]10− with CeIII. Owing to the existence of the stable 1 ∶ 2 complex, the solution equilibria are more complicated than for the α1 system (for which no 1 ∶ 2 complex has yet been detected) and evidence for two dimers is presented. The formation of labile complexes with proline has been detected by NMR.
Experimental
Materials
All chemicals were reagent grade and used as supplied. Water was purified by passage through a purification train. The following lacunary anion salts were prepared according to published methods and were identified by infrared spectroscopy and 31P NMR: α2-K10P2W17O61,12 α2-Li10P2W17O61,13 and α-Na12P2W15O56.12Synthesis of (NH4)7[Ce(P2W17O61)]·nH2O
This complex was first isolated as a minor by-product from the synthesis of [Ce4(OH2)9(OH)2(P2W16O59)2]14−.14 More efficient syntheses were subsequently developed.The products of the three preparations gave identical P-NMR spectra. Found: N, 1.94; H, 1.44; Ce, 3.04; P, 1.17; W, 63.11; Cl, 0.10; K, 0.20%. Calc. for (NH4)6.75K0.25[Ce(P2W17O61)]·0.27H2O·0.15NH4Cl: N, 1.96; H, 1.66; Ce, 2.84; P, 1.26; W, 63.40; Cl, 0.10; K, 0.20%. IR (ν/cm−1): 1085 (s), 1055 (w), 1024 (w), 1016 (w) 943 (s), 923 (s), 918 (s), 812 (vs), 783 (s). CeIV/III redox potential (literature8): 0.654 V (0.651 V) vs. Ag/AgCl.
Crystal structure determination
Single crystal X-ray analyses were performed on a Bruker-Siemens SMART CCD single crystal diffractometer equipped with an Mo-Kα anode and graphite monochromator (λ = 0.71073 Å). The orange crystal (0.17 × 0.12 × 0.04 mm) was mounted on a glass fiber under mineral oil to prevent water loss, and placed in a nitrogen stream at 173(2) K. The final unit cell was calculated using a least squares refinement of reflections culled from the entire data set. The data was corrected for Lorentz and polarization effects. An empirical absorption correction based on equivalent reflections15 was applied as incorporated in the SADABS program.16 All structures were solved using direct methods and refined against F2 using the routines included in the SHELXTL-PC software suite.17 Nitrogen atoms of ammonium cations were modeled as oxygen atoms because the former could not be distinguished from the latter. K, P, W and Ce atoms were refined anisotropically; oxygen atoms were refined isotropically. Hydrogen atoms were not included in the refinement model. (NH4)13K[{Ce(P2W17O61)}2](H2O)18, M = 9204.95, monoclinic, a = 46.1124(6), b = 12.7451(1), c = 26.4624(2) Å, β = 93.570(1), U = 15522.0(3), T = 173(2) K, space group C2/c, Z = 4, μ(Mo-Kα) = 25.825 mm−1, 84148 reflections measured, 18856 unique (Rint = 0.1610) which were used in all calculations. The final wR(F2) was 0.1594.CCDC reference number 169242.
See http://www.rsc.org/suppdata/dt/b1/b105967j/ for crystallographic data in CIF or other electronic format.
Physical measurements
Phosphorus-31 NMR was measured on a Varian Mercury 300 MHz spectrometer (P resonance frequency 121.472 MHz) interfaced to a Sun Microsystems workstation with the following parameters: spectral width, 10000 Hz; acquisition time, 6.4 s; pulse delay, 5 s; pulse width, 34.1°. All spectra were referenced to external 85% H3PO4, and the temperature was controlled by a VT temperature control unit. Tungsten-183 NMR spectra were recorded on a Bruker AM 300WB spectrometer (W resonance frequency 12.504 MHz), equipped with an Aspect 3000 computer with the following parameters: spectral width, 8064.516 Hz; acquisition time, 1.0158 s; pulse delay, 0.5 s; pulse width 45° tip angle. All spectra were referenced to external 2 M Na2WO4. Infrared spectra were recorded with a Nicolet FT-7000 spectrometer as KBr pellets. Cyclic voltammetry was measured at ambient temperature on a BAS100A system. A glassy carbon working electrode (diameter 3mm), a platinum wire counter electrode and an Ag/AgCl reference electrode (3M NaCl) were used. Approximate formal potential values (E1/2) were calculated from the cyclic voltammograms as the average of the cathodic and anodic peak potentials for each corresponding oxidation and reduction wave.P-NMR Titration
Titration of α2-[P2W17O61]10− with Ce(NO3)3 was carried out as follows: A concentrated solution of α2-[P2W17O61]10− was prepared by dissolving ca. 7.20 g of α2-Li10P2W17O61·22H2O in 10.0 mL D2O, and 0.5 mL of the solution was placed in an NMR tube. To this solution, Ce(NO3)3 (1.50 M) in D2O was added in 5 or 10 μL increments, and the 31P-NMR spectrum was measured at 23 °C. On adding Ce(NO3)3, new peaks belonging to the [Ce(α2-P2W17O61)2]17− complex (4) appeared, until the ratio of α2-[P2W17O61]10− and Ce(NO3)3 reached 2 ∶ 1. Plotting the integration ratio change between the peaks due to α2-[P2W17O61]10− and [Ce(α2-P2W17O61)2]17− against the amount of Ce(NO3)3 added gave the exact concentration (1.50 M) of the α2-[P2W17O61]10− solution.Results and discussion
Synthesis
The complex was isolated as a mixed potassium–ammonium salt from the reactions involving the precursor anions, α2-[P2W17O61]10− or α-[P2W15O56]12− (methods 1 and 2, respectively).| α2-[P2W17O61]10− + Ce(NO3)3 (3 eq.) → [Ce(α2-P2W17O61)]7− (Method 1) |
| α-[P2W15O56]12− + WO42− (2 eq.) + Ce(NO3)3 (2 eq.) → [Ce(α2-P2W17O61)]7− (Method 2) |
The direct method (method 1) between the monovacant lacunary anion α-[P2W17O61]10− and Ce(NO3)3 gave excellent isolated yields (90%). The trivacant lacunary derivative α-[P2W15O56]12− also gave the desired product under appropriate conditions in the presence of additional tungstate, although excess amounts of Ce(NO3)3 were necessary. The formation of an oily phase during the synthesis and its disappearance upon addition of NH4Cl (method 1) suggested the importance of the counter ions for this synthesis. Attempts to optimize the synthesis for the correct (1 ∶ 1) stoichiometry were unsuccessful, and considerable amounts of the 1 ∶ 2 complex (4) were formed.
X-Ray structural analysis
As illustrated in Fig. 1, the complex exists in dimeric form (1) in the solid state. The asymmetric unit contains a 1 ∶ 1 [(H2O)4Ce(α2-P2W17O61)]7− unit, and the complete anion is generated by a center of symmetry. The linkage between the two 1 ∶ 1 units occurs in a head to head fashion, via two Ce–O–W bridges formed between each cerium atom and one terminal oxygen atom belonging to the W2O10 duplet of the adjacent α2-P2W17O61 anion. Each cerium atom is nine-coordinate (monocapped square antiprism), with four sites occupied by one anion unit (acting as a tetradentate ligand), four by water molecules, and the ninth site by a terminal oxygen of the other anion. The average Ce–O bond length for the bonds formed with the ligand‘s oxygens in the lacuna (2.48 Å) is essentially equal to the sum of the Ce–O ionic radii for an 8-coordinate Ce3+ and 2-coordinate O2− (2.49 Å)18 and to the average Ce–O bond length in the [Ce(α2-P2W17O61)2]17− complex (2.46 Å).19 The bond length for the Ce–O bond joining the two halves of the anion (Ce–O2A, 2.57 Å) is comparable to the overall average Ce–OH2 bond distance (2.56 Å) and to the sum of the ionic radii for a 9-coordinate Ce3+ and 2-coordinate O2− (2.55 Å).18 The W–O framework of the anion does not show unusual features and the W–O and P–O distances are consistent with previous crystallographic reports of complexes formed with the α2-P2W17O61 anion.19,20![(left) Polyhedral representation of [Ce(α2-P2W17O61)(H2O)4]214− in the ammonium salt (white circles: cerium, grey circles: water oxygens) and (right) ball-and-stick representation of selected atoms (phosphorus atom is black).](/image/article/2002/DT/b105967j/b105967j-f1.gif) | ||
| Fig. 1 (left) Polyhedral representation of [Ce(α2-P2W17O61)(H2O)4]214− in the ammonium salt (white circles: cerium, grey circles: water oxygens) and (right) ball-and-stick representation of selected atoms (phosphorus atom is black). | ||
NMR analysis
The P-NMR spectrum of a solution of the isolated ammonium salt (20 mM in D2O) exhibits two pairs of resonances, A1 (−17.61 ppm), A2 (−14.57 ppm), and much weaker B peaks at −14.81 and −14.06 ppm, see Fig. 2(a). In order to understand this spectrum a series of measurements was made on solutions of Li10[α2-P2W17O61] with increasing amounts of Ce(NO3)3, see Fig. 3. Four resonances are observed when 0 < Ce/P2W17 < 0.5, two for unreacted [P2W17O61]10− at −6.70 (P1) and −13.39 ppm (P2) and two for the 1 ∶ 2 complex [Ce(P2W17O61)2]17− at −13.90 and −13.93 ppm. The 1 ∶ 2 complex is the only detectable species present when Ce/P2W17 = 0.5. When 0.5 < Ce/P2W17 < 1.0 the two NMR peaks gradually transform into the B resonances and the A resonances appear and increase in intensity. These results demonstrate that the species responsible for the B resonances is in fast exchange with the 1 ∶ 2 complex, but not with the species responsible for the A resonances. That “A” and “B” are connected by a slower exchange process was shown by 2D (EXSY) spectroscopy (see ESI). The relative intensities of the A and B peaks at Ce/P2W17 = 1.0 are concentration dependent, see Fig. 2(b) and (c), and are consistent with a “monomer(A)–dimer(B)” equilibrium, characterized by K = 1.46 ± 0.04 M−1 and ΔH = −18.2 ± 0.8 kJ mol−1, (Fig. 4 and ESI). Although it is tempting to identify B with the dimer revealed by the crystal structure, this does not explain the fast exchange with the 1 ∶ 2 complex and slow exchange with the monomer, [Ce(P2W17O61)(H2O)x]7−.21 We therefore propose equilibria involving four species, as shown in Scheme 1—the crystal dimer (1) and the “monomer” (2) in fast exchange, the 1 ∶ 2 complex (4) and an “ion-pair” of 4 with Ce3+ (3) also in fast exchange, and a “slow” exchange equilibrium between 3 and 1, 2. This scheme is further supported by the following W-NMR data.![P-NMR spectra of [Ce(α2-P2W17O61)]7−: (a) 20 mM of isolated complex at 23.0 °C (40 scans), (b) 150 mM at 23.0 °C (8 scans), (c) 15 mM at 23.0 °C (128 scans), and (d) 150 mM at 66.8 °C (8 scans) of in situ-prepared complex {[α2-P2W17O61]10−
+ Ce(NO3)3}.](/image/article/2002/DT/b105967j/b105967j-f2.gif) | ||
| Fig. 2 P-NMR spectra of [Ce(α2-P2W17O61)]7−: (a) 20 mM of isolated complex at 23.0 °C (40 scans), (b) 150 mM at 23.0 °C (8 scans), (c) 15 mM at 23.0 °C (128 scans), and (d) 150 mM at 66.8 °C (8 scans) of in situ-prepared complex {[α2-P2W17O61]10− + Ce(NO3)3}. | ||
![Chemical shift variation during the titration of α2-[P2W17O61]10− with Ce(NO3)3 (open circles and squares: B2 and B1 resonances, closed circles and squares: A2 and A1 resonances). Broken lines denote stoichiometry of the 1 ∶ 2 and 1 ∶ 1 complexes.](/image/article/2002/DT/b105967j/b105967j-f3.gif) | ||
| Fig. 3 Chemical shift variation during the titration of α2-[P2W17O61]10− with Ce(NO3)3 (open circles and squares: B2 and B1 resonances, closed circles and squares: A2 and A1 resonances). Broken lines denote stoichiometry of the 1 ∶ 2 and 1 ∶ 1 complexes. | ||
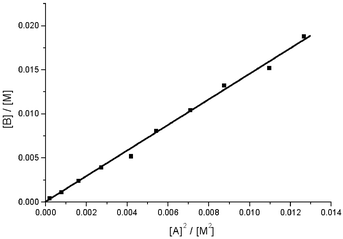 | ||
| Fig. 4 Plot of the integrated intensity of the B1 resonance versus the square of the intensity of the A1 resonance in D2O at 23.0 °C. The sample was prepared by mixing Li10(α2-P2W17O61) and Ce(NO3)3 in a 1 ∶ 1 ratio (initial concentration 150 mM). The slope (monomer–dimer equilibrium constant) is 1.46 ± 0.04 M−1 (R2 = 0.998). | ||
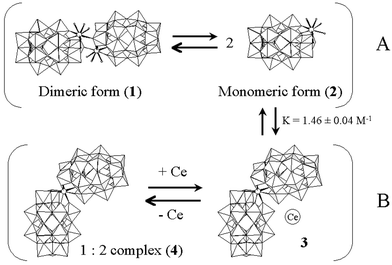 | ||
| Scheme 1 Suggested equilibria in D2O. Those represented by horizontal arrows are labile on the NMR timescale. | ||
The W-NMR spectrum of a saturated solution of the ammonium salt (∼20 mM 1) shows eight resonances (2 ∶ 2 ∶ 2 ∶ 1 ∶ 2 ∶ 2 ∶ 2 ∶ 2), Fig. 5(a). Attempts to generate a more soluble lithium salt by ion-exchange led to the formation of large amounts of the 1 ∶ 2 complex [spectrum shown in Fig. 5(e)], but the spectrum of the ammonium salt could be reproduced from an equimolar mixture of Li10[α2-P2W17O61] and Ce(NO3)3, Fig. 5(b). The “missing” line in this spectrum is assigned to W1,W2 [Fig. 1(b)], which are anticipated to have very different chemical shifts in a static dimer structure, but which become equivalent under monomer–dimer exchange at a rate, resulting in an undetectable coalesced line. Evidence for the intermediate exchange rate, is also provided by the broad A1 resonance (from the phosphorus adjacent to the cerium atoms) in the P-NMR spectrum, which narrows as the temperature is increased, Fig. 2(b) and (d).
![W-NMR spectra (see Table 1 for chemical shift values) of (a) [Ce(α2-P2W17O61)]7− (isolated complex ca. 20 mM), 230000 scans, (b) in situ-prepared [Ce(α2-P2W17O61)]7− (50 mM Li10(α2-P2W17O61) +
{50 mM Ce(NO3)3}, 40000 scans, (c) (b) +
DL-proline (20 eq.), 44000 scans, (d) (b) +
L-proline (20 eq.), 52000 scans, (e) lithium salts of [Ce(α2-P2W17O61)2]17− (1 g in 3 mL D2O), 46000 scans. The broadness of the lowest fieldline is attributed to slow rotational interconversion
of syn and anti isomers of the complex [see ref. 5(a)].](/image/article/2002/DT/b105967j/b105967j-f5.gif) | ||
| Fig. 5 W-NMR spectra (see Table 1 for chemical shift values) of (a) [Ce(α2-P2W17O61)]7− (isolated complex ca. 20 mM), 230000 scans, (b) in situ-prepared [Ce(α2-P2W17O61)]7− (50 mM Li10(α2-P2W17O61) + {50 mM Ce(NO3)3}, 40000 scans, (c) (b) + DL-proline (20 eq.), 44000 scans, (d) (b) + L-proline (20 eq.), 52000 scans, (e) lithium salts of [Ce(α2-P2W17O61)2]17− (1 g in 3 mL D2O), 46000 scans. The broadness of the lowest fieldline is attributed to slow rotational interconversion of syn and anti isomers of the complex [see ref. 5(a)]. | ||
| a Splitting of virtual “doublet” resulting from loss of mirror symmetry in the AA complex. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| a | 121 | −135 | −174 | −176 | −209 | −210 | −215 | −226 | |
| (2W) | (2W) | (2W) | (1W) | (2W) | (2W) | (2W) | (2W) | ||
| b | 120 | −136 | −176 | −177 | −210 | −211 | −216 | −227 | |
| 2W | 2W | 2W | 1W | 2W | 2W | 2W | (2W) | ||
| c | 304 | 118 | −135 | −172 | −175 | −209 | −215 | −224 | |
| (2W) | (2W) | (2W) | (2W) | (1W) | (4W) | (2W) | (2W) | ||
| d | 304 | 118 | −135 | −172 | −175 | −208 | −210 | −215 | −223 |
| (2W, 33 Hza) | (2W, 29 Hza) | (2W, 31 Hza) | (2W, 19 Hza) | (1W) | (2W, 6 Hza) | (2W) | (2W) | (2W) | |
| e | 323 | 145 | −136 | −171 | −179 | −211 | −213 | −217 | −226 |
| (2W, br.) | (2W) | (2W) | (2W) | (1W) | (2W) | (2W) | (2W) | (2W) | |
We have shown elsewhere that amino acids (AA) form weak complexes with [Ce(α1-P2W17O61)(H2O)x]7− and that these may be used to discriminate between the enantiomers of the heteropolyanions.10 When D,L-proline is added to a solution containing [Ce(α2-P2W17O61)(H2O)x]7− both P- and W-NMR spectra are modified, the A1 resonance is narrowed21 and the “missing” tungsten resonance is observed [Fig. 5(c)]. If L-proline is added, several of the W resonances are split as a result of the loss of mirror symmetry in the AA complex.22 These results imply that attachment of the amino acid to the monomer impedes the monomer–dimer exchange. The formation constant of the proline–polyanion complex was estimated to be 4.5 ± 0.1 M−1 from 1H NMR spectra (ESI). This value is comparable to those found for the corresponding complexes of the α1 isomer10 and for AA complexes of EuIII chelates.23
Conclusions
Isolation of the “1 ∶ 1” complex (1, in dimeric form) of Ce3+ and the α2 isomer of [P2W17O61]10− was achieved by the use of a 3-fold excess of Ce3+. Solutions of 1, or an equimolar mixture of Ce3+ and [P2W17O61]10−, are involved in equilibria linked to the stable 1 ∶ 2 complex. This behavior stands in contrast to that of mixtures of Ce3+ and the α1 isomer of [P2W17O61]10− which yield only a 1 ∶ 1 complex in rapid exchange with the dimeric form.10 There is no convincing evidence to date to suggest that 1 ∶ 2 complexes of any trivalent lanthanides with α1-[P2W17O61]10− can be formed, and this has been attributed to steric factors.24,25 Addition of proline to solutions of 1 prevents exchange between monomer and dimer forms by binding to the Ce3+ centre. The formation constant of the proline complex is comparable to that found for other amino acid complexes of lanthanides.Acknowledgements
This work was supported in part by the US Department of Energy (grant no.: DE-EF07-96ER14695), National Science Foundation (CHE9727417), Mitsubishi Chemical Co., and the Department of Chemistry at Georgetown University. The authors would like to thank Dr Knut Wassermann and Dr Michael H. Dickman for helpful discussions, and expert technical suggestions. We are grateful to Prof. Robert E. Bachman for training in and assistance with X-ray crystallography, and Prof. Angel C. de Dios and Dr Veeradej Chynwat for assistance with NMR spectroscopy.References and notes
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin, 1983 Search PubMed; (b) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef; (c) Y. Izumi, K. Urabe and M. Onaka, Zeolite, Clay, and Heteropoly Acid in Organic Reaction, Kodansha, Tokyo, 1992, 99 Search PubMed; (d) C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407 CrossRef CAS; (e) T. Okumura, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113 Search PubMed; (f) Chem. Rev., special edition on polyoxometalates, ed. C. L. Hill; 1998, 98 Search PubMed; (g) R. Neumann, Prog. Inorg. Chem., 1998, 47, 317 Search PubMed.
- A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239 CrossRef.
- C. M. Flynn, Jr. and G. D. Stucky, Inorg. Chem., 1969, 8, 333; C. M. Flynn, Jr. and G. D. Stucky, Inorg. Chem., 1969, 8, 335 CrossRef.
- R. D. Peacock and T. J. R. Weakley, J. Chem. Soc. A, 1971, 1836 RSC.
- (a) J. Bartis, S. Sukal, M. Dankova, E. Kraft, R. Kronzon, M. Blumenstein and L. C. Francesconi, J. Chem. Soc., Dalton Trans., 1997, 1937 RSC; (b) Q.-H. Luo, R. C. Howell, M. Dankova, J. Bartis, C. W. Williams, W. DeW. Horrocks, Jr., V. G. Young, Jr., A. L. Rheingold, L. C. Francesconi and M. R. Antonio, Inorg. Chem., 2001, 40, 1894 CrossRef CAS.
- (a) D. A. Malikov, M. S. Milyukova and E. V. Kuzovkina, Sourem. Metody Razd. I Opred. Radioaktiv. Elem., M., 1989, 99 Search PubMed from Zh. Khim., 1990, abstr. no. 1V233; D. A. Malikov, M. S. Milyukova and E. V. Kuzovkina, Chem. Abstr., 1990, 112, 241827m Search PubMed; (b) N. Y. Kremlyakova, A. P. Novikov, B. F. Myasoedov and N. V. Katargin, J. Radioanal. Nucl. Chem., 1990, 145, 183 CAS; (c) M. S. Milyukova, D. A. Malikov, E. V. Kuzovkina and B. F. Myasoedov, J. Radioanal. Nucl. Chem., 1986, 104, 81 CAS; (d) Y. M. Kulyakov, T. I. Trofimov, V. Y. Frenkel, I. A. Lebedev and B. F. Myasoedov, J. Anal. Chem. (Transl. of Zh. Anal. Khim.), 1981, 36, 1691 Search PubMed; (e) Y. M. Kulyako, I. A. Lebedev, V. Y. Frenkel, T. I. Trofimov and B. F. Myasoedov, Sov. Radiochem. (Engl. Transl.), 1981, 23, 671 Search PubMed; (f) N. P. Molochnikova, V. Y. Frenkel, B. F. Myasoedov and I. A. Lebedev, Sov. Radiochem. (Engl. Transl.), 1982, 24, 250 Search PubMed; (g) N. P. Molochnikova, V. Y. Frenkel and B. F. Myasoedov, J. Radioanal. Nucl. Chem., 1988, 121, 409; (h) M. S. Milyukova, N. S. Varezhkina and B. F. Myasoedov, Sov. Radiochem. (Engl. Transl.), 1990, 32, 361 Search PubMed; (i) N. P. Molochnikova, V. Y. Frenkel and B. F. Myasoedov, Radiokhimiya, 1989, 31, 65 Search PubMed; (j) J. M. Adnet, P. Brossard and J. Bourges, Proc. Int. Conf. Technol. Expo. Future Nucl. Syst.: Emerging Fuel Cycles Waste Disposal Options 1993, 1993, 2, 1008 Search PubMed; J. M. Adnet, P. Brossard and J. Bourges, Chem. Abstr., 1994, 120, 146793d Search PubMed; (k) A. M. Fedoseev and I. G. Tananaeuv, Radiokhimiya, 1994, 36, 422 Search PubMed.
- J. P. Ciabrini and R. Contant, J. Chem. Res.(M), 1993, (S)391, (M)2720 Search PubMed.
- N. Haraguchi, Y. Okaue, T. Isobe and Y. Matsuda, Inorg. Chem., 1994, 33, 1015 CrossRef CAS.
- M. Sadakane, M. H. Dickman and M. T. Pope, Angew. Chem., Int. Ed., 2000, 36, 2914 CrossRef CAS.
- M. Sadakane, M. H. Dickman and M. T. Pope, Inorg. Chem., 2001, 40, 2715 CrossRef CAS.
- Q. H. Luo, R. Howell, J. Bartis, M. Dankova, W. DeW. Horrocks and L. Francesconi, Pacifichem 2000, Honolulu, HI, Dec. 14–19, 2000 Search PubMed abstr. INOR 1078.
- R. Contant, Inorg. Synth., 1990, 27, 104 CAS.
- J. Bartis, Y. Kunina, M. Blumenstein and L. C. Francesconi, Inorg. Chem., 1996, 35, 1497 CrossRef CAS.
- A. Ostuni and M. T. Pope, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 199 CrossRef CAS.
- R. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33 CrossRef.
- G. M. Sheldrick, SADABS, Absorption Correction Method, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL 97, Program for the Refinement of Crystal Structures, release 97–2, Bruker Analytical Instruments, Madison, WI, USA, 1997 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751.
- A. Ostuni, M. Sc. Thesis, Georgetown University, Washington D. C., USA, 1998 Search PubMed.
- V. N. Molchanov, L. P. Kazanskii, E. A. Torchenkova and V. I. Simonov, Sov. Phys. Crystallogr. (Engl. Transl.), 1979, 24, 96 Search PubMed.
- The peak-widths at half-height of [Ce(α2-P2W17O61)]7− (ca. 10 mM in D2O at 22 °C) were 6.29 Hz and 3.06 Hz for the peaks at −17.40 and −14.44 ppm, respectively. In the presence of 16 eq. of L-proline, we observed two peaks at −17.56 and −14.59 ppm with peak-widths at half-height of 3.78 and 2.80 Hz, respectively.
- No line-splitting is observed in the presence of D,L-proline, since the (enantiomeric) adducts interconvert rapidly on the NMR timescale.
- (a) J. Kido, Y. Okamoto and H. G. Brittain, J. Org. Chem., 1991, 56, 1412 CrossRef CAS; (b) R. Hulst, N. K. De Vries and B. L. Feringa, J. Org. Chem., 1994, 59, 7453 CrossRef CAS.
- J. Bartis, M. Dankova, J. J. Lessmann, Q.-H. Luo, W. DeW. Horrocks, Jr. and L. C. Francesconi, Inorg. Chem., 1999, 38, 1042 CrossRef CAS.
- However, 1 ∶ 2 complexes with U4+ and Th4+, and the mixed-ligand species [Ce(α1-P2W17O61)(α2-P2W17O61)]17− have been isolated and structurally characterized, see ref. 19 and A. Ostuni, R. E. Bachman, A. K. Jameson and M. T. Pope, manuscipt in preparation.
Footnote |
| † Electronic supplementary information (ESI) available: representative P-31 EXSY spectrum for a solution of “1 ∶ 1” complex. Plot of lnKvs. 1/T for the equilibrium species A (1, 2) and B (3, 4). Determination of formation constant of proline complex; plot of inverse cerium-induced chemical shift of Hα versus [proline]/[1]. See http://www.rsc.org/suppdata/dt/b1/b105967j/ |
| This journal is © The Royal Society of Chemistry 2002 |