Americium(III) and europium(III) solvent extraction studies of amide-substituted triazine ligands and complexes formed with ytterbium(III)
Nathalie
Boubals
b,
Michael G. B.
Drew
*a,
Clément
Hill
b,
Michael J.
Hudson
a,
Peter B.
Iveson
a,
Charles
Madic
b,
Mark L.
Russell
a and
Tristan G. A.
Youngs
a
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, UK RG6 6AD. E-mail: m.g.b.drew@reading.ac.uk
bCommissariat à l'Energie Atomique, Valrhô, DEN/DRCP/SCPS/LCSE, Bât 399, B.P. 17171, 30207, Bagnols-sur-Cèze, Cedex, France
First published on 3rd December 2001
Abstract
4-Amino-bis(2,6-(2-pyridyl))-1,3,5-triazine L4 and several, amide derivatives with hydrophobic alkyl substituents have been synthesised. Solvent extraction studies carried out on Am(III) and Eu(III) with L4 and its amide derivatives in synergistic combination with α-bromodecanoic acid, show that these ligands can selectively extract actinides with respect to lanthanides. The structures of [H2L4]·2Cl·2.5H2O and two amide derivatives have been determined and show respectively the trans, trans; cis, cis; and cis, trans conformations of the adjacent aromatic rings. These observed conformations are in agreement with the results of quantum mechanics calculations on L4 and its protonated derivatives. The structures of two Yb complexes with amide derivatives are also reported with stoichiometry [Yb(L)(NO3)(H2O)4]·2NO3·0.5H2O and [Yb(L)(NO3)3(H2O)]·2MeCN and show the metal in 9 coordinate environments.
Introduction
One possible future scenario in nuclear reprocessing is the conversion or transmutation of the long-lived minor actinides, such as americium, into short-lived isotopes by irradiation with neutrons.1 In order to achieve this transmutation it is necessary to separate the trivalent minor actinides from the trivalent lanthanides by solvent extraction, because the lanthanides absorb neutrons effectively and hence prevent neutron capture by the transmutable actinides.For many years we have been designing and testing ligands for the co-extraction2,3 of lanthanides and actinides from nuclear waste and their subsequent separation.4,5 Oligoamines such as 2,2′:6′2″-terpyridine4–6 (L1) and 2,4,6-tris(2-pyridyl)-1,3,5-triazine5 (L2) have been shown to selectively extract actinides in preference to the lanthanides from nitric acid solutions into an organic phase. For extraction it proved necessary to use these ligands in synergistic combination with α-bromodecanoic acid. Separation factors for Am(III) relative to Eu(III) were found to be around 7 and 10 for L1 and L2 respectively.5 Although a separation factor of around 12 can be obtained with a more hydrophobic derivative of L2 (2,4,6-tris(4-tert-butyl-2-pyridyl)-1,3,5-triazine (L3), the synthesis is difficult and the ligand can only be prepared on a small scale. The promising solvent extraction results obtained with L2 and L3 led us to the synthesis of 4-amino-bis(2,6-(2-pyridyl))-1,3,5-triazine (L4). This ligand contains the same major binding cavity as L2 but is much more easily functionalised to prepare the appropriate hydrophobic derivatives. In this study, several new amide derivatives of L4 have been prepared containing either 2,2,4-trimethyl-1-pentyl (L5), 2,2-dimethyl-1-propyl (L6), cyclohexyl (L7), or heptyl (L8) as the hydrophobic alkyl substituent (Fig. 1). The Am(III)/Eu(III) separation–extraction performance of L4, L5, L7 and L8 was measured in combination with α-bromodecanoic acid. The structures of a bis hydrochloride salt of L4 and of the free ligands L5 3,5,5-trimethylhexanoylamino-bis(2,6-(2-pyridyl)-1,3,5-triazine) and L7 (4-cyclohexanoylamino-bis(2,6-(2-pyridyl))-1,3,5-triazine) together with the corresponding Yb(III) complexes of L6 and L7 were also determined.
 | ||
| Fig. 1 Structures of ligands. | ||
Experimental
Synthesis
L4 was prepared as described previously.7 3,5,5-Trimethylhexanoyl chloride, tert-butylacetyl chloride, cyclohexane carbonyl chloride, octanoyl chloride and ytterbium nitrate pentahydrate (99.9%) were used as received from Aldrich. Pyridine and acetonitrile were dried over 4 and 3 Å molecular sieves respectively.Preparation of ligands
Preparation of metal complexes
Solvent extraction studies
Aqueous solutions (800 μL) of diluted nitric acid (0.02 mol L−1 ≤ [HNO3] ≤ 0.13 mol L−1), spiked with radioisotopes 241Am and 152Eu, were contacted for 30 minutes by means of an automatic vortex shaker with organic solutions (800 μL), containing either ligand L4, L5, L7 or L8 ([L]initial = 0.02 mol L−1), diluted in a mixture of hydrogenated tetrapropene (TPH) and α-bromodecanoic acid ([αBrC10]initial = 1 mol L−1). Aqueous and organic solutions were mixed in 2 mL Nalgene tubes thermostatted at 22 °C. After phase separation by centrifugation, 500 μL samples of both phases were analysed using a gamma counting spectrometer (HPGe detector, Eurisys Mesures). The peaks at 59.54 and 121.78 keV were used for 241Am and 152Eu activity measurements, respectively. The concentration of nitric acid in the aqueous phase at equilibrium ([HNO3]eq) was determined by automatic titration with NaOH.The distribution ratio DM for a metallic cation M is defined as the ratio of the concentration of the metallic species in the organic phase at equilibrium over its concentration in the aqueous phase at equilibrium. The error of the measure of DM (M = Am(III) or Eu(III)) was estimated to be within 5%. The separation factor SFM1/M2 for two metallic cations M1 and M2 is defined as the ratio of their distribution ratios. The error of the determination of SFAm/Eu was estimated to be 7%.
Crystallography
The structures of the salt [H2L4]·2Cl·2.5H2O, the ligands L5·2H2O and L7, and the ytterbium complexes with L6 and L7 were determined. Crystal data and refinement details are provided in Table 1. Data for all 5 crystals were collected with Mo-Kα radiation using the MAR research Image Plate System. The crystals were positioned 70 mm from the image plate. 95 frames were measured at 2° intervals with a counting time of 2 min. Data analysis was carried out with the XDS program.8 Default refinement details are described here while differences for specific structures are included below. Structures were solved using direct methods with the SHELX86 program.9 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms on the carbon atoms and nitrogen atoms were included in calculated positions and given thermal parameters equivalent to 1.2 times those of the atom to which they were attached. Hydrogen atoms on water molecules were included when they could be located in a difference Fourier map and refined with distance constraints. The assignment of the positions of the nitrogen atoms in the pyridine rings was made straightforwardly on the basis of thermal parameters, dimensions, R values and hydrogen bond positions. An empirical absorption correction was made for the two metal complexes using the DIFABS program.10| Compound | [H2L4]·2Cl·2.5H2O | L7 | L5·2H2O | [Yb(L7)(NO3)(H2O)4]·2NO3·0.5H2O | [Yb(L6)(NO3)3(H2O)]·2MeCN | |
| Empirical formula | C13H17Cl2N6O2.5 | C20H20N6O | C22H29N6O3 | C20H29N9O14.5Yb | C24H28N11O10Yb | |
| Formula weight | 368.23 | 360.42 | 425.51 | 800.56 | 803.61 | |
| Temperature/K | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | |
| Crystal system, space group | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Monoclinic, P21/c | Monoclinic, P21/a | Monoclinic, C2/c | Monoclinic, C2/c | |
| a/Å | 10.158(12) | 9.946(12) | 12.458(14) | 24.35(3) | 8.935(14) | |
| b/Å | 13.890(15) | 17.15(2) | 14.997(17) | 16.760(18) | 12.887(14) | |
| c/Å | 14.249(15) | 11.849(14) | 12.515(14) | 14.812(17) | 15.004(17) | |
| α/° | 69.20(1) | (90) | (90) | (90) | 71.69(1) | |
| β/° | 67.61(1) | 109.12(1) | 93.54(1) | 103.49(1) | 87.14(1) | |
| γ/° | 71.60(1) | (90) | (90) | (90) | 78.96(1) | |
| Volume/Å3 | 1699 | 1910 | 2334 | 5878 | 1610 | |
| Z, Calculated density/Mg m−3 | 2, 1.439 | 4, 1.254 | 4, 1.211 | 4, 1.809 | 2, 1.658 | |
| Absorption coefficient/mm−1 | 0.404 | 0.082 | 0.083 | 3.266 | 2.974 | |
| F(000) | 764 | 760 | 908 | 3184 | 798 | |
| Reflections collected | 5890 | 6387 | 7430 | 6980 | 5198 | |
| Unique reflections/Rint | 5890 | 3383/0.0310 | 4416/0.0761 | 4420/0.0762 | 5198 | |
| Data/restraints/parameters | 5890/15/457 | 3383/0/245 | 4416/4/297 | 4420/0/190 | 4198/2/428 | |
| Final R indices [I > 2σ(I)] | R1 | 0.0479 | 0.0844 | 0.0920 | 0.1119 | 0.0296 |
| wR2 | 0.1284 | 0.2336 | 0.2439 | 0.2809 | 0.0712 | |
| R indices (all data) | R1 | 0.0722 | 0.1544 | 0.2214 | 0.2547 | 0.0367 |
| wR2 | 0.1419 | 0.2846 | 0.3061 | 0.3363 | 0.0755 | |
| Largest diff. peak and hole/e Å−3 | 0.260, −0.272 | 0.360, −0.233 | 0.697, −0.277 | 3.540, −1.922 | 0.997, −1.596 | |
In the structure of the salt [H2L4]·2Cl·2.5H2O, there are two formula units in the asymmetric unit. All the hydrogen atoms on the water molecules were located in a difference Fourier map and included in the refinement with distance constraints. The structure of L7 contained no solvent. L5 contained two water molecules in the asymmetric unit but the hydrogen atoms on these solvent molecules were not located. For [Yb(L7)(NO3)(H2O)4]·2NO3·0.5H2O the hydrogen atoms on the water molecules were not located. The data were of poor quality and only the metal atom was refined anisotropically. For [Yb(L6)(NO3)3(H2O)]·2MeCN the hydrogen atoms on the water molecules were located and refined with distance constraints. All structures were refined on F2 till convergence using SHELXL.11
CCDC reference numbers 172878–172882.
See http://www.rsc.org/suppdata/dt/b1/b104181a/ for crystallographic data in CIF or other electronic format.
Theoretical calculations
There are three possible conformations for L4 which can be characterised by the N–C–C–N torsion angles as tt (trans, trans), ct (cis, trans) and cc(cis, cis). We have analysed these conformations for L4, [HL4]+ and [H2L4]2+ using the Gaussian94 program.12 Starting models were built using the CERIUS2 software13 and the three rings were made approximately coplanar but no symmetry was imposed. Structures were then optimised using the 6-31G** basis set.Results and discussion
Synthesis
Hydrophobic amide derivatives of 4-amino-bis(2,6-(2-pyridyl))-1,3,5-triazine L4 were synthesised by reaction of L4 with the appropriate acid chloride in refluxing pyridine. This method has been used previously for the acylation of 2,4-diamino-s-triazines.14 There is a wide choice of hydrophobic substituents because of the large number of commercially available acid chlorides. The two ligands containing 2,2-dimethyl-1-propyl and cyclohexyl alkyl groups (L6 and L7), were prepared in order to facilitate crystallisation of the corresponding lanthanide complexes. The more organo-soluble L5 and L8 were prepared for the solvent extraction experiments. The 2,2,4-trimethyl-1-pentyl alkyl chain in L5 was chosen because it bears a closer resemblance to the TPH solvent used as the organic phase in the extraction experiments. TPH (hydrogenated tetrapropene) is an industrial aliphatic diluent containing highly branched alkanes.Extraction studies
The synergistic extraction of trivalent actinides and lanthanides observed when combining tridentate planar ligands such as L2 with α-bromodecanoic acid (αBrC10) in TPH can be described by the following equilibrium:5| M3+ + 3αBrC10 + L ⇌ ML(αBrC10)3 + 3H+ |
We have studied the synergistic extraction of Am(III) and Eu(III) by combining ligands L4, L5, L7 or L8 with α-bromodecanoic acid, used here as a cationic exchanger. The replacement of nitrate anions by α-bromodecanoate anions enhances the extraction of [ML]3+ complexes in TPH. The results of these experiments are summarised in Table 2 together with the results related to ligands L2 and L3 and are also shown in Fig. 2 for ligands L4 and L5, and Fig. 3 for ligands L7 and L8, respectively.
| Ligand | [HNO3]eq | D Eu(III) | D Am(III) | SFAm/Eu |
|---|---|---|---|---|
| a Organic solution: [αBrC10]initial = 1 M, [L]initial = 0.02 mol L−1, TPH. Aqueous solution: [HNO3]initial = variable, T = 22 °C. | ||||
| L1 | 0.03 | 2.8 | 27 | 9.5 |
| 0.05 | 0.3 | 3.5 | 10 | |
| 0.08 | 0.09 | 0.8 | 9 | |
| 0.11 | 0.03 | 0.3 | 9 | |
| L2 | 0.03 | 8.7 | 124 | 14 |
| 0.05 | 0.9 | 12 | 14 | |
| 0.08 | 0.2 | 2.1 | 11 | |
| 0.11 | 0.08 | 0.8 | 9 | |
| L4 | 0.03 | 4.6 | 45 | 9.5 |
| 0.05 | 0.5 | 5.5 | 10 | |
| 0.08 | 0.08 | 0.7 | 9.5 | |
| 0.11 | 0.02 | 0.2 | 11.5 | |
| L5 | 0.02 | 2.2 | 14 | 6.5 |
| 0.04 | 0.5 | 4.8 | 9.5 | |
| 0.06 | 0.12 | 1.1 | 9 | |
| 0.09 | 0.04 | 0.39 | 9 | |
| 0.13 | 0.02 | 0.21 | 9 | |
| L7 | 0.03 | 2.2 | 24 | 11 |
| 0.05 | 0.24 | 2.8 | 12 | |
| 0.10 | 0.04 | 0.4 | 10 | |
| L8 | 0.03 | 2.4 | 19 | 8 |
| 0.06 | 0.3 | 3.5 | 12 | |
| 0.10 | 0.03 | 0.4 | 12 | |
![Am(iii) and Eu(iii) extraction by L4 and L5 in a synergistic mixture with α-bromodecanoic acid. Organic solution: [αBrC10]initial
= 1 M, [L]initial
= 0.02 mol L−1, TPH. Aqueous solution: [HNO3]initial
= variable, T
= 22 °C. The symbols denote each measurement of DM. The approximately horizontal lines represent calculated SF values.](/image/article/2002/DT/b104181a/b104181a-f2.gif) | ||
| Fig. 2 Am(III) and Eu(III) extraction by L4 and L5 in a synergistic mixture with α-bromodecanoic acid. Organic solution: [αBrC10]initial = 1 M, [L]initial = 0.02 mol L−1, TPH. Aqueous solution: [HNO3]initial = variable, T = 22 °C. The symbols denote each measurement of DM. The approximately horizontal lines represent calculated SF values. | ||
![Am(iii) and Eu(iii) extraction by L7 and L8 in a synergistic mixture with α-bromodecanoic acid. Organic solution: [αBrC10]initial
= 1 M, [L]initial
= 0.02 mol L−1, TPH. Aqueous solution: [HNO3]initial
= variable, T
= 22 °C. The symbols denote each measurement of DM. The approximately horizontal lines represent calculated SF values.](/image/article/2002/DT/b104181a/b104181a-f3.gif) | ||
| Fig. 3 Am(III) and Eu(III) extraction by L7 and L8 in a synergistic mixture with α-bromodecanoic acid. Organic solution: [αBrC10]initial = 1 M, [L]initial = 0.02 mol L−1, TPH. Aqueous solution: [HNO3]initial = variable, T = 22 °C. The symbols denote each measurement of DM. The approximately horizontal lines represent calculated SF values. | ||
Although not as efficient as ligand L2, ligands L4, L5, L7 and L8 appeared to be as efficient as ligand L1 when used in a synergistic mixture with α-bromodecanoic acid for the extraction of Am(III) and its separation from Eu(III). For the three rather hydrophobic substituted ligands L5, L7 and L8, the log–log plots of the experimental DM values vs. [HNO3]eq fitted well with a linear regression. The slope of which was close to −3, in agreement with the above proposed extraction equilibrium. However, for the non-substituted ligand L4, the log–log plots of the experimental DM values vs. [HNO3]eq were better fitted with a linear regression, the slope of which is close to −4, possibly because of the redistribution of this rather hydrophilic ligand in the aqueous acidic phase after protonation (i.e.: [HNO3]eq ≥ 0.08 M).
Structural studies
| Symmetry elements. For [H2L4]·2Cl·2.5H2O: $1 x, y, 1 + z; $2 1 − x, 1 − y, −z; $3 −x − 1, 2 − y, −z; $4 −x − 1, 2 − y, 1 − z. For L7: $1 x, 0.5 − y, 0.5 + z. For L5·2H2O: $3 0.5 + x, 0.5 − y, z; $6 2.5 − x, 0.5 + y, 1 − z; $7 x + 0.5, 0.5 − y, z − 1. For [Yb(L7)(NO3)(H2O)4]·2NO3·0.5H2O: $1 x, 1 − y, z − .5; $2 0.5 − x, 0.5 − y, 1 − z; $4 1 − x, y, 0.5 − z; $5 1 − x, 1 − y, 1 − z; $6 x, 1 − y, 0.5 + z. For [Yb(L6)(NO3)3]·2MeCN: $4 1 − x, 1 − y, −z; $5 x + 1, y, z. | |||
|---|---|---|---|
| [H2L4]·2Cl·2.5H2O | |||
| O(100)⋯Cl(1) | 3.15 | N(11B)⋯O(101)$3 | 2.69 |
| O(100)⋯Cl(2)$2 | 3.11 | N(11B)⋯N(25B) | 2.70 |
| O(101)⋯Cl(3) | 3.17 | N(27A)⋯O(100)$2 | 2.84 |
| O(101)⋯Cl(4)$4 | 3.14 | N(27A)⋯Cl(1) | 3.26 |
| O(102)⋯Cl(1) | 3.07 | N(27B)⋯Cl(3)$3 | 3.33 |
| O(103)⋯Cl(3) | 3.21 | N(27B)⋯Cl(3) | 3.34 |
| O(103)⋯Cl(4) | 3.21 | N(31A)⋯O(102) | 2.68 |
| O(104)⋯Cl(4) | 3.13 | N(31A)⋯N(23A) | 2.72 |
| O(104)⋯Cl(2)$1 | 3.11 | N(31B)⋯O(103) | 2.69 |
| N(11A)⋯N(25A) | 2.71 | N(31B)⋯N(23B) | 2.69 |
| N(11A)⋯Cl(2) | 3.09 | ||
| L7 | |||
| N(41)⋯N(11)$1 | 3.00 | ||
| L5·2H2O | |||
| N(11)⋯N(41)$3 | 2.94 | O(100)⋯O(200)$7 | 2.91 |
| O(43)⋯O(200) | 2.79 | O(100)⋯O(200)$6 | 2.96 |
| O(200)⋯N(31) | 2.84 | ||
| [Yb(L7)(NO3)(H2O)4]·2NO3·0.5H2O | |||
| O(100)⋯O(61)$4 | 2.79 | O(102)⋯O(64)$5 | 2.67 |
| O(100)⋯O(62)$4 | 2.96 | O(102)⋯O(51)$1 | 2.86 |
| O(101)⋯O(44)$6 | 2.97 | N(41)⋯O(54)$2 | 2.82 |
| O(101)⋯O(61)$5 | 3.07 | ||
| [Yb(L6)(NO3)3]·2MeCN | |||
| O(100)⋯N(500)$5 | 2.76 | N(41)⋯N(400)$4 | 3.14 |
![The structure of the [H2L4]2+ cation A showing the hydrogen bond pattern (dotted lines). Ellipsoids at 30% probability.](/image/article/2002/DT/b104181a/b104181a-f4.gif) | ||
| Fig. 4 The structure of the [H2L4]2+ cation A showing the hydrogen bond pattern (dotted lines). Ellipsoids at 30% probability. | ||
![The structure of the [H2L4]2+ cation B showing the hydrogen bond pattern (dotted lines). Ellipsoids at 30% probability.](/image/article/2002/DT/b104181a/b104181a-f5.gif) | ||
| Fig. 5 The structure of the [H2L4]2+ cation B showing the hydrogen bond pattern (dotted lines). Ellipsoids at 30% probability. | ||
The structure of L7 is shown in Fig. 6. There is one strong hydrogen bond between N(41) and N(11) (x, 0.5 − y, 0.5 + z) with an N⋯N distance of 3.00 Å. The arrangement of the three nitrogen atoms is cis, cis (cc). On the other hand, the structure of L5·2H2O exhibits the ligand in the ct conformation as shown in Fig. 7. A water molecule O(200) forms donor hydrogen bonds to both N(31) at 2.84(1) and O(43) at 2.79(1) Å. The second water molecule in the asymmetric unit (not shown in the Figure) forms two hydrogen bonds to O(200) but not to L5. In addition N(11) forms an intermolecular hydrogen bond to N(41) (5 + x, 0.5 − y, z) at 2.94 Å.
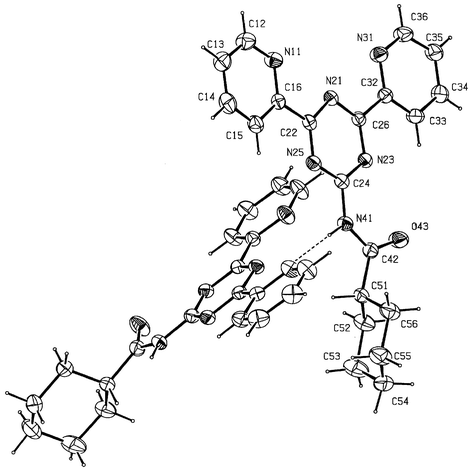 | ||
| Fig. 6 The structure of L7 with ellipsoids at 30% probability. | ||
 | ||
| Fig. 7 The structure of L5·2H2O with ellipsoids at 30% probability. One water molecule, present in the asymmetric unit, is not shown. | ||
The fact that L5 is in the ct conformation and L7 is in the cc conformation (and indeed that [H2L4]2+ is in the tt conformation) is particularly interesting. Our theoretical calculations on the parent amine (see below) show that there are only small energy differences between these three conformations, much lower than is found for ligands (e.g. 2,2′:6′2″-terpyridine (L1))6 with a central pyridine ring. The energy barrier between conformations is also likely to be much lower in these ligands containing a central triazine ring than in those containing a central pyridine ring where the ortho-hydrogen atoms on adjacent rings restrict rotation.
The structure of the [Yb(L7)(NO3)(H2O)4]2+ cation is shown in Fig. 8. There are two nitrate anions together with a disordered water molecule in the asymmetric unit. The structure of the cation shows the Yb to be in a 9-coordinate environment with the metal bond lengths shown in Table 4, together with the atomic numbering scheme. The water molecules show the shortest bonds although there is significant variation [2.29(2)–2.39(2) Å] but in addition, each of them forms two strong hydrogen bonds with adjacent non-coordinated nitrate anions. This formation of a doubly charged ionic complex containing a lanthanide with only one nitrate ion is unique and is not found in any of the 100 or so structures that we have determined of lanthanide nitrate complexes with terdentate nitrogen ligands.4,6,15 The bond to the central triazine nitrogen atom Yb(1)–N(21) is at 2.39(2) Å much shorter than the bonds to the outer pyridine nitrogen atoms (Yb(1)–N(31) 2.52(2), Yb(1)–N(11) 2.57(2) Å). The amide N–H group is hydrogen bonded to a nitrate oxygen atom (which is not bonded to the metal) from an adjacent molecule, viz (N(41)⋯O(54) (0.5 − x, 0.5 − y, 1 − z), 2.84 Å. There are many hydrogen bonds (Table 3) between the coordinated water molecules and nitrates in adjacent molecules.
| [Yb(L7)(NO3)(H2O)4]·2NO3·0.5H2O | |||
| Yb(1)–O(102) | 2.290(16) | Yb(1)–O(42) | 2.437(19) |
| Yb(1)–O(101) | 2.313(15) | Yb(1)–N(31) | 2.52(2) |
| Yb(1)–O(103) | 2.385(18) | Yb(1)–O(41) | 2.548(18) |
| Yb(1)–N(21) | 2.39(2) | Yb(1)–N(11) | 2.57(2) |
| Yb(1)–O(100) | 2.398(16) | ||
| [Yb(L6)(NO3)3(H2O)]·2MeCN | |||
| Yb(1)–O(41) | 2.277(4) | Yb(1)–N(21) | 2.419(4) |
| Yb(1)–O(100) | 2.298(4) | Yb(1)–O(52) | 2.470(4) |
| Yb(1)–O(51) | 2.362(4) | Yb(1)–N(11) | 2.489(5) |
| Yb(1)–O(61) | 2.400(4) | Yb(1)–N(31) | 2.519(5) |
| Yb(1)–O(62) | 2.404(4) | ||
![The structure of the [Yb(L7)(NO3)(H2O)4]2+ cation with the atomic numbering scheme. Ellipsoids at 30% probability. Hydrogen atoms on the water molecules could not be located and are not shown.](/image/article/2002/DT/b104181a/b104181a-f8.gif) | ||
| Fig. 8 The structure of the [Yb(L7)(NO3)(H2O)4]2+ cation with the atomic numbering scheme. Ellipsoids at 30% probability. Hydrogen atoms on the water molecules could not be located and are not shown. | ||
This structural type, [Yb(L7)(NO3)(H2O)4]2+, is different from the two types obtained previously for Yb(III) and L4 as in [Yb(L4)(NO3)2(H2O)2], [NO3], Yb is coordinated to one tridentate L4 ligand, two bidentate nitrates and two water molecules and in [Yb(L4)(NO3)3(H2O)], Yb is coordinated to one L4, two bidentate nitrates, one monodentate nitrate and one water molecule.15 The latter structure is similar to that of [Yb(L6)(NO3)3(H2O)] which is shown in Fig. 9. This structure also contains two solvent acetonitrile groups in the asymmetric unit. This structure is also 9-coordinate as one of the nitrate anions is monodentate. As is usually the case, the shortest Yb–O bond is to the monodentate nitrate Yb(1)–O(41) 2.277(4) Å, followed by the bond to the water molecule Yb(1)–O(100) 2.298(4) Å. The next shortest bonds are those to the bidentate nitrate although the bond to O(52) at 2.470(4) Å is significantly longer than the other three (2.362(4), 2.400(4), 2.404(4) Å). As was found for [Yb(L7)(NO3)(H2O)4]·2NO3·0.5H2O, the bond to the triazine nitrogen atom is, at 2.419(4) Å, significantly shorter than the bonds to the other nitrogen atoms (2.489(5), 2.519(5) Å). The amide nitrogen atom in this case is hydrogen bonded to a solvent acetonitrile molecule (N(41)⋯N(400) (1 − x, 1 − y, −z) 3.14 Å).
![The structure of the [Yb(L6)(NO3)3(H2O)] complex with ellipsoids at 30% probability. One of the two solvent acetonitrile molecules is shown.](/image/article/2002/DT/b104181a/b104181a-f9.gif) | ||
| Fig. 9 The structure of the [Yb(L6)(NO3)3(H2O)] complex with ellipsoids at 30% probability. One of the two solvent acetonitrile molecules is shown. | ||
We ascribe no particular significance to the difference in stoichiometry between the two metal complexes of Yb with L6 and L7. In previous work we have shown that there can be very different coordination geometries for specific metals with these planar terdentate ligands and it seems likely that the replacement of one bidentate and one monodentate nitrate by three water molecules is not unusual and that both structures together with others are likely to co-exist in solution. The only thing in common is that in both cases the metal cations are 9-coordinate as is usually found for ytterbium. It is likely that with these ligands the smaller lanthanides are 9-coordinate and have the form [ML(NO3)n(H2O)m]p+ with n = 1, 2 or 3; m = 6 − 2n + x, where x is the number of monodentate nitrates. General structural trends show that the number of monodentate nitrates will be either 1 or 0, and that at least one bidentate nitrate will be coordinated so that the charge p on the complex is 0, 1 or 2. It can also be considered that there are several different entities for the complexes in solution.
It is interesting to note that 2,6-bis(5,6-dipropyl-1,2,4-triazin-3-yl)pyridine which provides a separation ratio for An/Ln of greater than 100,16 when used without a synergist, forms the 1 ∶ 3 complex in the presence of nitrate with the smaller lanthanides (Sm–Lu)17 but not for the larger lanthanides (La–Sm).18 This suggests that ligands that always form 1 ∶ 1 complexes are not likely to give high separation ratios in the absence of a synergist. While we have only determined crystal structures with Yb and not the other lanthanides, all indications from previously obtained structural information2,4,15 is that when Yb forms 1 ∶ 1 complexes with particular ligands then so do the other lanthanides and therefore we conclude that these ligands do not form 1 ∶ 3 complexes with any lanthanide. Another factor that may lessen the usefulness of L4 and derivatives is that they always form hydrogen bonds with either solvent molecules or other metal complexes, a feature not present in the ML33+ complex of 2,6-bis(5,6-dipropyl-1,2,4-triazin-3-yl)pyridine, which has a hydrophobic exterior. The propensity for hydrogen bond formation as indicated by Table 3 is widespread in all the crystal structures for ligands and metal complexes alike and the aggregation of molecules may occur in solution to prevent efficient extraction.
Theoretical analysis of L4, [HL4]+ and [H2L4]2+
Results are summarised in Table 5 for L4, [HL4]+ and [H2L4]2+. For the neutral ligand L4, the relative energies of the cc, ct and tt form were 2.36, 0.00, and 0.49 kcal mol−1 respectively. This result contrasts with that found for terpyridine where the relative energies of the three forms were 12.55, 6.90, and 0.00 kcal mol−1 respectively.6 There is a much smaller difference in energy in L4 for the three conformers compared to terpyridine because there is no possible clash of ortho hydrogen atoms as the central ring is a triazine rather than a pyridine. It can be argued that the differences between the energies of the three conformers in L4 are less than packing effects, which may account for the occurrence of the ct and tt forms in the crystal structures of L5 and L7 respectively (Figs. 7 and 6). It is likely that these conformational preferences in L5 and L7 will be comparable to those of L4. This is in contrast to terpyridine where the energy differences between conformers are much more significant such that only the tt form is likely to be observed. In general it can be noted from the Cambridge Crystallographic Database that all polypyridine structures exhibit the trans conformation in the absence of a metal or another coordinating species. While our calculations have been carried out in the gas phase, it seems likely that the similar qualitative results will pertain in solution, that it will be more favourable for terdentate ligands with central triazine rings to form the cc conformation necessary for metal complexation than those with a central pyridine ring.| Conformation | Protonated nitrogen | cc | ct | tt |
|---|---|---|---|---|
| L4 | 2.36 | 0.00 | 0.49 | |
| L2 | 12.55 | 6.90 | 0.00 | |
| [HL4]+ | N(11) | 9.77 | 14.67 | 14.55 |
| N(31) | ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(11) N(11) |
12.22 | N(11) |
|
| N(21) | 0.00 | 10.13 | 22.19 | |
| N(25) | 28.98 | 27.07 | 14.55 | |
| N(23) | N(25) |
14.05 | N(25) |
|
| N(27) | 51.10 | 46.15 | 44.38 | |
| [H2L4]2+ | N(11), N(21) | 24.35 | 40.53 | 34.20 |
| N(11), N(31) | 5.37 | 1.79 | 0.00 | |
| N(11), N(25) | 33.99 | 39.75 | 45.83 | |
| N(11), N(23) | 24.55 | 14.18 | 13.82 | |
| N(11), N(27) | 41.70 | 44.15 | 45.70 | |
| N(21), N(23) | N(21), N(25) |
34.00 | N(21), N(25) |
|
| N(21), N(25) | 37.82 | 50.57 | 49.71 | |
| N(21), N(27) | 50.98 | 60.49 | 73.44 | |
| N(21), N(31) | N(11), N(21) |
18.52 | N(11), N(21) |
|
| N(25), N(31) | N(11), N(23) |
26.33 | N(11), N(23) |
|
| N(23), N(25) | 69.04 | 51.03 | 35.82 | |
| N(23), N(27) | N(25), N(27) |
91.29 | N(25), N(27) |
|
We then investigated the possible structures of [HL4]+ cations. Given that N(11) and N(31) are equivalent as indeed are N(25) and N(23) in cc and tt conformations we constructed 14 different structures, combinations of the nitrogen to be protonated and the three possible conformations. The cc conformation with N(21) protonated was ca. 10 kcal mol−1 lower in energy than all the others. The relative stability of this configuration is probably due to a combination of factors, N(21) is the preferred nitrogen to be protonated, and also it can form weak hydrogen bonds to the ortho nitrogen atoms in the terminal pyridine rings. In addition, there are no steric repulsions owing to adjacent ortho hydrogen atoms. The next most favourable configuration is the cc conformation with N(11) protonated but this only forms one weak intramolecular hydrogen bond. The ct conformation with N(21) protonated is disfavoured owing to repulsion between ortho hydrogen atoms on N(21) and C(33). It is interesting that in this case (and in others) the presence of two mutually ortho-hydrogen atoms leads to a twist in the rings of ca. 33–46° leading to a loss of conjugation. The highest energies (>44 kcal mol−1) occur with N(27) protonated even though this does not lead to increased steric repulsions so that these energies must be due to the unfavourable nature of protonation at that atom.
Results for [H2L4]2+ are less clear cut and show three configurations with comparable low energies within 5.4 kcal mol−1, all with N(11) and N(31) protonated. The lowest energy configuration has the tt conformation in which there are two weak hydrogen bonds formed between N(11)–H⋯N(25) and N(31)–H⋯N(23) and no ortho⋯ortho interactions. This is the structure found in the crystal structure of [H2L4]·2Cl·2.5H2O (Figs. 4 and 5) where the configuration is further stabilized by intermolecular hydrogen bonds. The other conformations ct and cc also have low energies. With a cis conformation the pyridine nitrogen atom N(11) and/or N(31) can form hydrogen bonds to the central nitrogen atom N(21) but again there are no undesirable ortho⋯ortho interactions. All other configurations have an energy of 13.8 kcal mol−1 greater than the minimum. There are several features that can be discerned from the energy distribution. Unlike in [HL4]+ protonation of N(21) is not favourable, because the second proton, wherever placed, always leads to ortho⋯ortho intramolecular interactions unless it is situated on N(27) which is an unfavourable site. Protonation on N(25) {or N(23)} is favourable in the trans conformation because it gives rise to N(25)–H⋯N(11) or {N(23)–H⋯N(31)} interactions but not in the cis conformation because of ortho⋯ortho interactions which lead to rotation of the rings. An intermediate situation arises in configurations such as the cis conformation with N(11) protonated where there is a weak hydrogen bond between N(11)–H and N(21). However, a repulsion between N(25)–H and C(13)–H, which is resolved via an intermediate rotation of 20° compared to the ca. 40°, found where there is no such hydrogen bond formation. We can draw the overall conclusion that the order of preference for protonation is N(11) > N(21) > N(25) > N(27) but that this order can be varied according to which other nitrogen atoms are protonated and the presence of solvent which can lead to intramolecular hydrogen bonds. The energy barrier to rotation between cis and trans in L4 with a central triazine ring is far less than that observed with a central pyridine ring as in terpy where ortho⋯ortho hydrogen interactions provide a high energy barrier.
Conclusion
The experimental extraction studies indicate that these new amide ligands have selectivities towards Am(III) that are comparable to that of ligand L2. Furthermore, since they can be synthesised on a large scale much more easily than ligand L2, they are promising reagents in industrial solvent extraction for the separation of An(III) and Ln(III). The solid state studies show that these ligands form 1 ∶ 1 complexes with the lanthanides of a similar type to those formed with the parent L4. These ligands however show a propensity to form intermolecular hydrogen bonds through the amide groups which may prevent more efficient extraction. These ligands do not form the 1 ∶ 3 complexes found with 2,6-bis(5,6-dipropyl-1,2,4-triazin-3-yl)pyridine. The formation of this stoichiometry in a lanthanide complex might well be indicative of high separation factors as this ligand has a value for An/Ln of greater than 100.Acknowledgements
We are grateful for the financial support by the European Union Nuclear Fission Safety Programme Task 2 (Contracts F141-CT-96-0010 and F1KW-CT2000-00087). We would also like to thank the EPSRC and the University of Reading for funding of the image-plate system. The use of the Origin 2000 at the University of Reading High Performance Computer Centre (HPCC) is gratefully acknowledged.References
- Proceedings of the 5th OECD/NEA Information Exchange Meeting on Actinide and Fission Product Partitioning and Transmutation, Mol, Belgium, 1998, EUR 18898 EN Search PubMed.
- G. Y. S. Chan, M. G. B. Drew, M. J. Hudson, P. B. Iveson, J. O. Liljenzin, M. Skålberg, L. Spjuth and C. Madic, J. Chem. Soc., Dalton Trans., 1997, 649 RSC.
- P. B. Iveson, M. G. B. Drew, M. J. Hudson and C. Madic, J. Chem. Soc., Dalton Trans., 1999, 3605 RSC.
- M. G. B. Drew, P. B. Iveson, M. J. Hudson, J. O. Liljenzin, L. Spjuth, P. Y. Cordier, Å. Enarsson, C. Hill and C. Madic, J. Chem. Soc., Dalton Trans., 2000, 821 RSC.
- P. Y. Cordier, C. Hill, P. Baron, C. Madic, M. J. Hudson and J. O. Liljenzin, J. Alloys Compd., 1998, 271, 738 CrossRef.
- M. G. B. Drew, M. J. Hudson, P. B. Iveson, M. L. Russell, J. O. Liljenzin, M. Skålberg, L. Spjuth and C. Madic, J. Chem. Soc., Dalton Trans, 1998, 2873 RSC.
- F. H. Case and E. Croft, J. Am. Chem. Soc., 1959, 81, 905 CrossRef CAS.
- W. Kabsch, J. Appl. Crystallogr., 1988, 21, 916 CrossRef CAS.
- SHELX86: G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 Search PubMed.
- N. Walker and D. Stuart, Acta Crystallogr., Sect. A, 1983, 39, 158 CrossRef.
- SHELXL: G. M. Sheldrick, Program for crystal structure refinement, University of Göttingen, 1993 Search PubMed.
- Gaussian94 (Revision A.1) , M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. A. Keith, G. A. Peterson, J. A. Montogomery, K. Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresham, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challalcombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. Gonzalez and J. A. Pople, Gaussian, Inc., Pittsburgh, PA, 1995 Search PubMed.
- CERIUS2 , Molecular Simulations Inc., San Diego, California, USA Search PubMed.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Kooijman and A. L. Spek, J. Org. Chem., 1996, 61, 6371 CrossRef CAS.
- M. G. B. Drew, M. J. Hudson, P. B. Iveson, C. Madic and M. L. Russell, J. Chem. Soc., Dalton Trans., 2000, 2711 RSC.
- Z. Kolarik, U. Müllich and F. Gassner, Ion Exch. Solvent Extr., 1999, 17, 23 Search PubMed.
- M. G. B. Drew, D. Guillaneux, M. J. Hudson, P. B. Iveson, C. Madic and M. L. Russell, Inorg. Chem. Commun., 2001, 4, 12 CrossRef CAS.
- M. G. B. Drew, D. Guillaneux, M. J. Hudson, P. B. Iveson and C. Madic, Inorg. Chem. Commun., 2001, 4, 462 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |