EPR and UV/VIS spectroscopic investigations of VO2+ complexes and compounds formed in alkali pyrosulfates
Søren B.
Rasmussen
,
K. Michael
Eriksen
and
Rasmus
Fehrmann
*
Department of Chemistry and ICAT (Interdisciplinary Research Center for Catalysis), Technical University of Denmark, DK-2800 Lyngby, Denmark
First published on 6th December 2001
Abstract
The catalytically important molten salt–gas system M2S2O7–M2SO4–V2O5/SO2(g) (M = Na, K, Rb, Cs) has been investigated by X- and Q-band EPR spectroscopy. In order to obtain information about the V(IV) complex formation in the melts, samples rather dilute in V2O5 were quenched from the molten state at 450–460 °C to 0 °C. EPR spectra of the quenched samples were recorded on samples with alkali to vanadium (M/V) ratios 40, 80 and 160. The spectra show that two V(IV) complexes dominate in the melt regardless of the type of alkali metal ion. In systems with low activity of sulfate a paramagnetic V(IV) complex with g∥ = 1.915, g⊥ = 1.978 and line widths 5–15 Gauss is observed. In systems saturated with M2SO4 the obtained EPR spectra show a paramagnetic complex with the g-tensors g∥ = 1.930, g⊥ = 1.980 and line widths 20–60 Gauss. These results fit very well with the assumption that the species VO(SO4)22− and SO42− are in equilibrium with VO(SO4)34−. It has also been shown for the system M2S2O7–M2SO4(sat)–V2O5/SO2(g) that the line widths in the system increase with higher cation radius, and depend linearly on the volume fraction of the sample occupied by the cation. This indicates that spin–spin relaxation effects are the major contribution to line broadening. Combining information from UV/VIS and EPR spectra shows that the VO2+ unit in the molten salt solvent exhibits electronic properties close to aqueous solutions of V(IV).
Introduction
The molten salt–gas system M2S2O7/M2SO4/V2O5–SO2/O2/SO3/N2 (M = Na, K and/or Cs) is generally accepted as a realistic model system of the sulfuric acid catalyst.1 Earlier work2 has shown that during catalyst working conditions, an equilibrium between V(IV) and V(V) is found in the catalyst dependent on the SO2/SO3 partial pressure ratio. Not much is generally known about the structure of vanadium complexes in pyrosulfate melts. However, recent multi-nuclear NMR investigations3,4 have shown that V(V) is present as slightly distorted octahedral complexes in the catalytically important concentration range (molar ratio M/V = 2–4). Particularly important has been the X-ray investigation5 of the compound Cs4(VO)2O(SO4)4 and the subsequent confirmation by NMR spectroscopy that the (VO)2O(SO4)44− complex exists in the molten state as well as in the solid. A recent Raman spectroscopic investigation6 on these systems is in accordance with these results. The dimeric anion (VO)2O(SO4)44− is believed to be the catalytically active component in the catalyst.7The paramagnetic ([Ar]3d1) vanadyl complex has been thoroughly investigated by EPR for years, however almost entirely in apolar organic solvents or water. The EPR spectra of most vanadyl complexes can be described by a spin Hamiltonian, H, including the electron-Zeeman interaction and the electron-vanadium nuclear hyperfine interaction (1):
| H = βH(gxxŜx + gyyŜy + gzzŜz) + hc(AxxŜxÎx + AyyŜyÎy + AzzŜzÎz) | (1) |
This paper presents the results of an EPR investigation on V(IV) complexes in M2S2O7–M2SO4–V2O5/SO2(g) (M = Na, K, Rb or Cs) melts at low vanadium concentrations and on precipitating V(IV) compounds at higher concentrations. Melts with low vanadium concentrations allow recording of well resolved EPR spectra on both molten and quenched samples leading to more detailed information on the nature of the complexes, whereas melts with higher concentrations allow the formation of sufficient amounts of crystals to record their powder EPR spectra.
Experimental
Chemicals
Alkali pyrosulfate was made by thermal decomposition of the corresponding peroxodisulfate (analytical grade) as earlier described.12 Cs2S2O8 and Rb2S2O8 are not commercially available and were synthesized as described for Cs2S2O8.13 It proved necessary to recrystallize Na2S2O8 before decomposition to obtain sufficient purity (>99.9%). V2O5 was from Cerac (Pure >99.9%) and used without any further treatment. SO2 was from AGA (>99.9%). All handling of the chemicals was performed in a dry air glove box with a typical water content less than 10 ppm.The M2S2O7–M2SO4(sat)–V2O5/SO2(g) system
Mixtures of M2S2O7 and M2SO4 were prepared by melting and mixing the components in a sealed ampoule. The solubility of sulfates in pyrosulfates is rather low,11,12e.g. the solubility of K2SO4 in K2S2O7 is around 4 mole % at 450 °C. In order to ensure sulfate saturation even after a possible coordination of sulfate to V(IV), a mole fraction of sulfate, X(M2SO4)![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 0.06 (of the binary system), was chosen, regardless of the alkali metal. Visual inspection of the molten samples confirmed excess sulfate as a white precipitate. V2O5 was added to this mixture at a M/V (M2S2O7/V2O5) molar ratio of 40. The chemicals were filled into a quartz ampule (diameter = 12 mm, 300 mm length) in the glove box and
sealed immediately under ca. 0.7 bar SO2. The sealed ampules were transferred to a home made quartz tubular furnace wound with kanthal wire and were equilibrated for at least 100 h above the melting point of the mixture, i.e. 450–460 °C dependent on the type of alkali metal. From the furnace the ampules were dropped directly into icy water. Several attempts to quench into liquid nitrogen proved less successful, probably due to slow heat transfer through the N2 gas film formed on the surface of the hot ampule. A too slow cooling of the samples resulted partly in formation of crystalline V(IV) compounds in the samples obscuring the EPR spectrum.
0.06 (of the binary system), was chosen, regardless of the alkali metal. Visual inspection of the molten samples confirmed excess sulfate as a white precipitate. V2O5 was added to this mixture at a M/V (M2S2O7/V2O5) molar ratio of 40. The chemicals were filled into a quartz ampule (diameter = 12 mm, 300 mm length) in the glove box and
sealed immediately under ca. 0.7 bar SO2. The sealed ampules were transferred to a home made quartz tubular furnace wound with kanthal wire and were equilibrated for at least 100 h above the melting point of the mixture, i.e. 450–460 °C dependent on the type of alkali metal. From the furnace the ampules were dropped directly into icy water. Several attempts to quench into liquid nitrogen proved less successful, probably due to slow heat transfer through the N2 gas film formed on the surface of the hot ampule. A too slow cooling of the samples resulted partly in formation of crystalline V(IV) compounds in the samples obscuring the EPR spectrum.
After quenching, the ampules were cut open in the glove box and some of the mixture ground and transferred to quartz capillary tubes (2.0 mm outer diameter, 0.5 mm wall thickness). The samples were sealed under vacuum. The capillary tubes were of unspecified quality, but tested negative for any EPR background signal (e.g. Fe(III)).
Samples with other M/V ratios were made by dilution of the M/V = 40 mixture with a premixed M2S2O7–M2SO4 mixture.
The M2S2O7–V2O5/SO2(g) system
Samples without SO42−-saturation are sensitive to the decomposition of the solvent melt, i.e. [eqn. (2)]| S2O72− ⇄ SO3 + SO42− | (2) |
which causes SO42− contamination of the M2S2O7. Only carefully selected batches of pyrosulfate of high purity were used. The mixing procedure was the same as in the previous case, except that the samples were not remelted and requenched after being transferred to the capillaries as in the case of the M2S2O7–M2SO4(sat)–V2O5/SO2(g) system. This was avoided in order not to decompose the pyrosulfate further. For the same reason no spectra of molten samples have been recorded. It was not possible to record well resolved spectra with M/V ratios smaller than 160, the spectra smeared out due to increased spin–spin relaxation.
Compound formation and isolation
The V(IV) compounds were synthesised in M2S2O7–V2O5 melts with the alkali–vanadium ratios M/V = 3–5 in a pyrex flow cell and washed out with water (except for the mixed valence V(IV)–V(V) salt K6(VO)4(SO4)8 which is rather water soluble and prepared otherwise14) as described in detail earlier. The gas composition used was 10% SO2, 11% O2 and 79% N2, corresponding to the synthesis gas in a traditional sulfuric acid plant. In the case of preparation of K6(VO)4(SO4)8, this gas was converted more than 90%, i.e. containing more than 9% SO3.EPR spectroscopy
The EPR spectra were recorded on a Bruker EMX spectrometer with a 12 kW 10″ magnet. Room temperature X-band spectra were recorded with a Bruker ER4102ST cavity. High temperature X-band spectra were recorded with a Bruker ER4114HT cavity whereas the room temperature Q-band spectra were obtained with a Bruker ER5106Q cavity.Selected EPR spectra were simulated using the Simpow15 computer program.
Spectrophotometric measurements
The optical spectrum was obtained using a Jasco 570 UV/VIS/NIR spectrophotometer. A sample of M2S2O7–V2O5/SO2(g) with M/V molar ratio of 40 was measured by placing the quartz capillary sample used for EPR measurements in the beam. Since a significant background signal was measured due to reflexions from the cylindrical quartz capillary tube the spectrum was fitted to a function with three gaussian terms and one constant (y0) [eqn. (3)], | (3) |
The spectrum was recorded in the range ν = 25000 cm−1 (400 nm) to 8000 cm−1 (1250 nm).
Results and discussion
The composition and structure of the V(IV) complexes in pyrosulfate melts are as mentioned not as well known as the V(V) complexes. Based on the knowledge of the V(V) complexes and a very recent investigation on V(IV) in pyrosulfate, the following reactions (4) and (5) will be considered for the rest of the discussion:| Dissolution: V2O5 + 2S2O72− ⇄ (VO)2O(SO4)44− | (4) |
| Reduction: (VO)2O(SO4)44− + SO2 ⇄ 2VO(SO4)22− + SO3 | (5) |
Furthermore, in presence of excess sulfate the V(IV) complex will coordinate one additional sulfate [eqn. (6)]:
| Complex reaction: VO(SO4)22− + SO42− ⇄ VO(SO4)34− | (6) |
At the applied SO2 pressure the equilibrium, eqn. (5), is almost completely shifted to the right, i.e. towards V(IV). A short V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and four equatorial oxygen ligands from two bidentate sulfate ions are expected for VO(SO4)22−, since unidentate sulfate groups would lead to an unlikely coordination number of three instead of five where the trans position may coordinate additional sulfate (unidentately) to form the hexa-coordinated VO(SO4)34−, which is coordinatively saturated. In VO(SO4)22− the trans position will be free or loosely coordinated to the solvent. This type of coordination is common for the vanadyl(IV) cation.
O bond and four equatorial oxygen ligands from two bidentate sulfate ions are expected for VO(SO4)22−, since unidentate sulfate groups would lead to an unlikely coordination number of three instead of five where the trans position may coordinate additional sulfate (unidentately) to form the hexa-coordinated VO(SO4)34−, which is coordinatively saturated. In VO(SO4)22− the trans position will be free or loosely coordinated to the solvent. This type of coordination is common for the vanadyl(IV) cation.
The M2S2O7–M2SO4(sat)–V2O5/SO2(g) system
Fig. 1 shows the EPR spectra of samples with M/V = 40, 80 and 160 recorded in the X- and Q-band. All spectra are recorded at room temperature after quenching from 450–460 °C to 0 °C, except for the samples with M/V = 160 which in addition are recorded in the X-band in the molten state at 450–460 °C. The Q-band spectra of the samples with M/V = 160 have been simulated using the Simpow computer program. | ||
| Fig. 1 X-band (A) and Q-band (B) EPR spectra of VO(SO4)34− in the M2S2O7–M2SO4(sat)–V2O5/SO2(g) system with M/V = 40, 80 and 160 (M = Na, K, Rb and Cs). All spectra are recorded at room temperature after quenching from 450–460 °C except for the indicated M/V = 160 samples recorded at 450–460 °C in the X-band. Simulated spectra are shown stipulated. | ||
The general EPR features of the quenched samples are quite similar with two general trends. The X- and Q-band spectra both show axial symmetry typical for octahedral VO2+ complexes with the hyperfine structure (due to coupling to the 51V nucleus, I = 7/2), best resolved in the Q-band spectra. The line width of the spectra increases with increasing vanadium concentration (lower M/V ratio) and with the molar weight of the alkali cation. The samples Rb/V = 40, Rb/V = 80, Cs/V = 40 and Cs/V = 80 could not be recorded with well resolved hyperfine structure in the X-band and are therefore not shown in Fig. 1.
The quenched spectra are typical immobilized axial symmetric spectra also known from, e.g., frozen vanadyl-containing Schiff bases.16,17 Axial symmetric spectra can be well characterized by g-tensors and hyperfine coupling constants, i.e. the parameters g⊥, g∥, a⊥, a∥ and one line width. The Q-band spectra, stipulated in Fig. 1, are computer simulations using these five parameters and assuming Lorentzian line shapes. The initial parameter sets for the iterations were obtained by an algorithm16 using the well-resolved Q-band spectra. No differences were found in the parameters dependent on the vanadium concentration (within experimental error), hence the best resolved spectrum for each alkali metal was used for the accurate calculation of the EPR parameters, regardless of the concentration. The final parameters of the simulations are listed in Table 1. It can be seen that the parameters for the quenched samples are identical within the experimental error of ±0.005 and ±10 for g and a respectively, i.e.g∥ = 1.930, g⊥ = 1.980, a∥ = 200 Gauss, a⊥ = 70 Gauss. Only the parameters of the parallel feature for the Cs-based samples deviate slightly. It should be stressed that observable in-plane anisotropy rarely occurs in EPR spectra, even though some rhombicity is present.
| M | g ⊥ | g ∥ | Δg∥/Δg⊥a | g 0 (450 °C) | g 0 (calc)b | a ⊥ | a ∥ | a 0 (450 °C) | a 0 (calc)c |
|---|---|---|---|---|---|---|---|---|---|
| a Δg∥/Δg⊥ = ge − g∥/ge − g⊥. b g 0(calc) = 1/3(2g⊥ + g∥). c a 0(calc) = 1/3(2a⊥ + a∥). | |||||||||
| Na | 1.982 | 1.930 | 3.36 | 1.970 | 1.966 | 71.7 | 200 | 110 | 115 |
| K | 1.980 | 1.930 | 3.29 | 1.970 | 1.963 | 70.7 | 202 | 108 | 114 |
| Rb | 1.980 | 1.930 | 3.24 | 1.967 | 1.963 | 67.1 | 194 | 108 | 109 |
| Cs | 1.981 | 1.940 | 3.16 | 1.968 | 1.966 | 65.3 | 187 | 108 | 106 |
However, the line width changes significantly by increasing size of the cation (Fig. 2). In order to discuss this, the line width for the M/V = 160 series has been plotted versus the void fraction (%), i.e. the volume fraction of the sample which is not occupied by the cation, 1 − (4 /3NA)Vm−1r3π, r being the ionic radius of the cation, NA Advogadro's number and Vm the molar volume of the sample. Since the samples are very dilute in vanadium, the molar volumes of the pure alkali pyrosulfates have been used.18 The molar volume of the solid has been extrapolated from the liquid state. Fig. 2 shows this dependency both in the solid state at room temperature using the well resolved perpendicular components and in the molten state at 450–460 °C using the isotropic line width. Table 1 summarizes the parameters. Both in the solid and the liquid state an excellent linear correlation (R2 > 0.95) is found, indicating that the line broadening can be explained solely by a spin–spin relaxation effect.
 | ||
| Fig. 2 Line width dependence on void fraction (see text) of the alkali metal cations M = Na, K, Rb, Cs in the M2S2O7–M2SO4(sat)–V2O5/SO2(g) system for samples with molar ratios M/V = 160 in the liquid state at 450 °C (open circles) and in the solid state at room temperature (filled circles). | ||
The high temperature X-band EPR spectra (Fig. 1A) of the molten samples with M/V = 160 exhibit eight isotropic lines of varying intensity as always found for monomeric vanadyl complexes in solution. The different alkali pyrosulfate solvents seem not to affect the first coordination sphere of the complex. Thus no variation in the isotropic g value or the isotropic hyperfine structure constant is observed as with respect to the type of the alkali cation. In some of the spectra, a skew baseline can be seen, probably due partly to formation of dimeric or polymeric V(IV) species where coupling along the chains may smear out the hyperfine structure. The measured isotropic parameters fit well with the calculated isotropic values from the quenched samples—considering the large temperature difference—as it can be seen in Table 1, indicating that we are dealing with the same complex in both phases.
The M2S2O7–V2O5/SO2 system
The X- and Q-band EPR spectra of the vanadyl complexes without sulfate saturation are shown in Fig. 3. The spectra represent axial symmetric complexes as in the sulfate saturated system, but the line width is much smaller, 5–15 G vs. 20–60 G for samples with sulfate saturation. This indicates that there is less distortion of the VO2+ unit for VO(SO4)22− compared to VO(SO4)34− complexes present in the sulfate saturated system. Furthermore, the EPR parameters of the parallel features are—not surprisingly—changed, as can be seen from Table 2. The value of g∥ is typically about 0.015 smaller in the case of VO(SO4)22− compared to VO(SO4)34−. The perpendicular components are less affected. This shows that the character of the VO bond is important for the relaxation path of the unpaired electron.
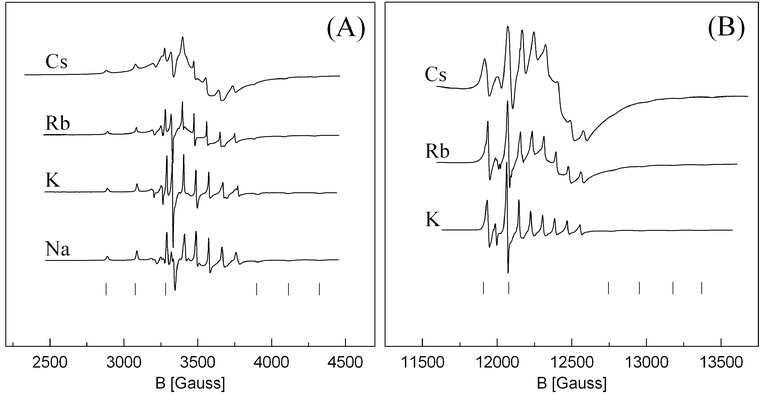 | ||
| Fig. 3 X-band (A) and Q-band (B) EPR spectra of VO(SO4)22− in the M2S2O7–V2O5/SO2(g) system with M/V = 160. All spectra are recorded at room temperature after quenching from 450–460 °C. Parallel features are indicated by “|” below the spectra. | ||
This system seems to show the same trend on line width versus alkali metal as the sulfate saturated system, and even more pronounced. However it was not possible to record well resolved spectra of the Cs based system for proper determination of line widths, even at Cs/V = 160. An attempt to increase the Cs/V ratio further did not increase the resolution significantly, but decreased the S/N ratio unacceptably. The Rb based system suffered to some extent from the same problems. Furthermore it was impossible to expose the samples to high temperature without decomposition of the complex, and in the case of M = Na decomposition occurred already at room temperature which made it impossible to obtain pure spectra of Na/V = 160 in Q-band. Consequently, it was not possible to construct a line width vs. void fraction plot.
Properties of VO2+ compounds crystallized from pyrosulfate melts
The g-values for seven different compounds synthesized in our laboratories19–21 have been determined by optimization using the Simpow program as shown in Table 3. The powder EPR spectra of several of the compounds have been given elsewhere. A varying degree of tetragonal distortion is found, judged from the g-anisotropy. These results show that EPR powder spectra of such compounds, which might appear isotropic from the immediate appearance of the spectra, actually exhibit anisotropy, which, however, becomes unresolved because of the relatively large line widths observed in these systems.| Compound | g ⊥ | g ∥ | g iso a | Ref. |
|---|---|---|---|---|
| a Only where applicable. b Mixed valence V(IV)–V(V) compound. c Unpublished. | ||||
| β-VOSO4 | 1.972 | 1.922 | 19 | |
| Na2VO(SO4)2 | 1.970 | 1.939 | 20 | |
| K4(VO)3(SO4)5 | 1.962 | 1.969 | 1.964 | 21 |
| K6(VO)4(SO4)8b | 1.978 | 1.930 | 1.972 | 14 |
| Rb2(VO)2(SO4)3 | 1.977 | 1.917 | c | |
| Cs2(VO)2(SO4)3 | 1.977 | 1.913 | c | |
| Na26(VO)5(SO4)18 | 1.980 | 1.925 | c | |
Electronic properties of VO2+ in pyrosulfate melts
According to the classic works22,23 on the electronic assignment of VO2+ complexes, the 3d1 electron occupies the dxy orbital in the ground state. Having a compressed octahedral geometry, the following simplified relations (7) and (8) between the paramagnetic and electronic transitions apply,17,24,25| Δg∥ = g∥ − ge = −8λβ1*2β2*2/Δ | (7) |
| Δg⊥ = g⊥ − ge = −2λβ2*2επ*2/δ | (8) |
λ being the spin–orbit coupling constant, where λ is estimated to be 170 cm−1 for the free V4+ ion, βi*2 and επ*2 the wavefunction coefficients, Δ = Ex2 − y2 − Exy and δ = Exz,yz − Exy are the energies for the electronic transitions between the indicated orbitals. The ratio Δg∥/Δg⊥ is very sensitive to tetragonal distortion—e.g. the strength of the vanadyl bond—of the octahedrons26 since it is directly related to the energy ratio Δ/δ. From the data in Tables 1 and 2 it seems that the sulfate saturated vanadyl complexes are less distorted (Δg∥/Δg⊥ = 3.2–3.5) than the sulfate free complexes (Δg∥/Δg⊥ = 3.5–3.7), which shows that the trans coordinated sulfate in the saturated samples weakens the VO2+ bond, as expected. These complexes, however, are more distorted than VO2+ on oxide surfaces (Δg∥/Δg⊥ = 1–3), but less distorted than VO2+ in silicate glasses (Δg∥/Δg⊥ = 3–4.4).27
The value of β2*2 can be obtained by equation (9):

| (9) |
where P is proportional to the expectation value of r−3 in the 3d orbitals of a free V4+ ion. A value of 184.5 Gauss is used in these calculations, as used by Sharma and co-workers.26
In Fig. 4, the optical spectrum of the complex present in the acidic quenched melt, VO(SO4)22−, is shown. The spectrum has been resolved in three gaussian components as shown by resolved curves and a constant contribution for background correction. The summarized function (10) is
| f(ν) = 1.56 + 0.89exp(−(ν − 11350)2/(2 × 11002)) + 0.72exp(−(ν − 14050)2/(2 × 19302)) + 18500exp(−(ν − 77000)2/(2 × 127802)). | (10) |
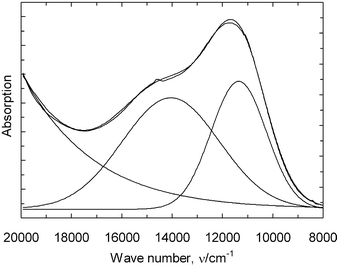 | ||
| Fig. 4 Absorption spectrum of the vanadyl complex VO(SO4)22− in the quenched M2S2O7–V2O5/SO2(g) system. | ||
The last term has no physical/chemical meaning, but provides a mathematical fit of the absorption from the charge transfer and strong V(V) bands in the UV region.
The transition assigned as Δ in eqn. 7 corresponds to dxy → dx2 − y2 and appears at 14050 cm−1 in VO(SO4)22−, while the transition assigned δ in eqn. (8) corresponds to dxy → dxz,yz, for the complex and is found at 11350 cm−1. In the region above 20000 cm−1, charge transfer bands well described17,22,28 for VO2+ are seen, but contributions from traces of V(V) complexes in this region cannot be ruled out, since V(V) is known to absorb strongly in this region. Using the estimated value for λ of 170 cm−1 together with the observed energies for the d–d transition band from the optical spectra and the observed g- and A-values from the EPR measurements on the quenched melts, the wave function coefficients can be estimated for the complex VO(SO4)22− in the frozen solution using eqns. (7)–(9).
This leads to the wave functions parameters, β1*2
= 1.028, β2*2
= 0.907 and επ*2
= 0.894. β1*2 characterize the in-plane σ-bonding of VO(SO4)22− and a value of one shows that there is no, or only very weak, covalent σ-bonding between the vanadium atom and the oxygen atoms in the equatorial ligand plane. The quantity 1 −
β2*2
= 0.093 corresponds to the fraction of unpaired d electrons delocalised over ligand orbitals. This indicates a weaker VO bond in this complex compared to VO(H2O)52+ and VO(acac)2 but a possible effect from packing of the vanadyl units in the Na2S2O7 structrure during quenching of the samples
can't be ruled out. The term (1 −
επ*2) = 0.106 indicates some influence of π-bonding between vanadium and the vanadyl oxygen not unlike VO(acac)2 which has chelating ligands such as VO(SO4)22− is likely to have.
All wave function parameters, however, are close to unity compared to for example VO2+ on oxide surfaces. This indicates that the electronic properties of the vanadyl unit is well preserved even in this molten salt with high ionic strength of the solvent compared to aqueous solutions. This also gains support from the very similar molar absorptivity found to be around 19.0 l mol−1 cm−1 for VO2+ formed in both aqueous and pyrosulfate solvents.2,27
Conclusions
It has been shown that two VO2+ complexes exist in M2S2O7–M2SO4–V2O5/SO2(g) (M = Na, K, Rb or Cs) melts in the molar ratio range M/V = 40–160. The spin Hamiltonian parameters have been derived, and it has been shown that line broadening of the EPR spectra of these complexes due to spin–spin relaxation is evident. The electronic properties of VO2+ in pyrosulfate melts have been discussed by calculating wave function parameters, β1*2, β2*2 and επ*2, using the obtained spin Hamiltonian parameters and energy levels obtained from an absorption spectrum of VO(SO4)22− in Na2S2O7.Acknowledgements
The authors wish to thank I. Laursen and K. Nielsen, Technical University of Denmark, for helpful discussions. NATO Science for Peace Programme (SfP 971984) has supported this investigation.References
- S. Boghosian, R. Fehrmann, N. J. Bjerrum and G. N. Papatheodorou, J. Catal., 1989, 119, 121 CrossRef CAS.
- D. A. Karydis, K. M. Eriksen, R. Fehrmann and S. Boghosian, J. Chem. Soc., Dalton Trans., 1994, 2151 RSC.
- O. B. Lapina, V. M. Mastikhin, A. A. Shubin, K. M. Eriksen and R. Fehrmann, J. Mol. Catal., 1995, 99, 123 Search PubMed.
- O. B. Lapina, V. V. Terskikh, A. A. Shubin, V. M. Mastikhin, K. M. Eriksen and R. Fehrmann, J. Phys. Chem., 1997, 101, 9188 CrossRef CAS.
- K. Nielsen, R. Fehrmann and K. M. Eriksen, Inorg. Chem., 1993, 32, 4825 CrossRef CAS.
- S. Boghosian, F. Borup and A. Chrissanthopoulos, Catal. Lett., 1997, 48, 145 Search PubMed.
- O. B. Lapina, B. S. Bal'zhinimaev, S. Boghosian, K. M. Eriksen and R. Fehrmann, Catal. Today, 1999, 51, 469 CrossRef CAS.
- V. M. Mastikhin, G. M. Polyakova, Ya. Zyulkovskii and G. K. Boreskov, Kinet. Katal, 1970, 11, 1463, (Engl. Transl., 1971, 11, 1219) Search PubMed.
- K. M. Eriksen, D. A. Karydis, S. Boghosian and R. Fehrmann, J. Catal., 1995, 155, 32 CrossRef CAS.
- K. M. Eriksen, R. Fehrmann and N. J. Bjerrum, J. Catal, 1991, 132, 263 CrossRef CAS.
- S. B. Rasmussen, K. M. Eriksen and R. Fehrmann, J. Phys. Chem, 1999, 103, 11282 CrossRef CAS.
- N. H. Hansen, R. Fehrmann and N. J. Bjerrum, Inorg. Chem., 1982, 21, 744 CrossRef CAS.
- G. E. Folkmann, G. Hatem, R. Fehrmann, M. Gaune-Escard and N. J. Bjerrum, Inorg. Chem., 1991, 30, 4057 CrossRef CAS.
- K. M. Eriksen, K. Nielsen and R. Fehrmann, Inorg. Chem., 1996, 35, 480 CrossRef CAS.
- M. Nilges, Program SIMPOW, Illinois ESR Research Center NIH, Illinois, IL, Division of Research Resources Grant No. RR0181 Search PubMed.
- N. D. Chasteen, in Biological Magnetic Resonance, ed. L. J. Berliner and J. Reubens, Plenum Press, New York, 1981, pp. 53 Search PubMed(please note misprints in formulas pp. 71–72).
- D. Kivelson and S-K. Lee, J. Chem. Phys., 1964, 41, 1896 CrossRef CAS.
- G. Hatem, F. Abdoun, M. Gaune-Escard, K. M. Eriksen and R. Fehrmann, Thermochim. Acta., 1998, 319, 33 CrossRef CAS.
- S. Boghosian, K. M. Eriksen, R. Fehrmann and K. Nielsen, Acta Chem. Scand., 1995, 49, 703 Search PubMed.
- R. Fehrmann, S. Boghosian, G. N. Papatheodorou, K. Nielsen and R. W. Berg, Inorg. Chem., 1991, 29, 3294.
- R. Fehrmann, S. Boghosian, G. N. Papathedorou, K. Nielsen and R. W. Berg, Inorg. Chem., 1989, 28, 1847 CrossRef.
- C. J. Ballhausen and H. B. Gray, Inorg. Chem., 1962, 1, 111 CrossRef CAS.
- C. K. Jørgensen, Acta Chem. Scand., 1957, 11, 73 Search PubMed.
- N. M. Atherton, in Principles of Electron Magnetic Resonance, ed. T. J. Kemp, Ellis Horwood, Chichester, 1993 Search PubMed.
- K. Dyrek and M. Che, Chem. Rev., 1997, 97, 305 CrossRef CAS.
- V. K. Sharma, A. Wokaun and A. Baiker, J. Phys. Chem., 1986, 90, 2715 CrossRef CAS.
- H. Farah, M. P. Brungs, D. J. Miller and G. R. Belton, Phys. Chem. Glasses, 1998, 39, 318 Search PubMed.
- J. Selbin, Coord. Chem. Rev., 1966, 1, 293 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |