Potentiometric and spectroscopic studies on transition metal complexes of GlyLys(Gly) and Asp-ε-Lys
Csilla Kállaya, Katalin Várnagya, Imre Sóvágó*a, Daniele Sannab and Giovanni Micerac
aDepartment of Inorganic and Analytical Chemistry, University of Debrecen, H-4010 Debrecen, Hungary
bIstituto C.N.R. per l‘Applicazione delle Tecniche Chimiche Avanzate ai Problemi Agrobiologici, Via Vienna 2, I-07100 Sassari, Italy
cDepartment of Chemistry, University of Sassari, Via Vienna 2, I-07100 Sassari, Italy
First published on 28th November 2001
Abstract
Copper(II), nickel(II) and zinc(II) complexes of the peptides GlyLys(Gly) and Asp-ε-Lys, containing the amide functions at the ε-amino groups of the lysyl residues were studied by potentiometric, UV-VIS and EPR spectroscopic methods. The stoichiometry of the major species formed in the copper(II)–GlyLys(Gly) system is [CuH−1L] and the EPR spectroscopic data indicate the existence of two isomeric forms of the complex. The nickel(II) and zinc(II)–GlyLys(Gly) systems have been characterised by the formation of stable [ML]+ complexes containing bis(NH2,CO) co-ordination and a macrochelate. Deprotonation and co-ordination of the amide groups were detected in the copper(II) and nickel(II) complexes. The co-ordination chemistry of Asp-ε-Lys is best described by the metal binding ability of the amino acid residues and it's high affinity for dimerisation. The stoichiometry of the dinuclear complexes can be given as [M2L2], containing only amino acid binding sites. Deprotonation and co-ordination of the amide functions were suggested only in the copper(II)-containing systems, resulting in the formation of the [Cu2H−2L2]2− dinuclear complex.
Introduction
Peptide–amide bonds of oligopeptides and proteins are generally built up from the α-carboxylic and amino groups of two amino acids in adjacent positions. The co-ordination chemistry of these peptides have already been thoroughly studied and the most important findings reviewed by several authors.1,2 Metal binding of these molecules is generally described via the co-ordination of the terminal amino group and, subsequently, the deprotonated amide nitrogens to form stable five-membered linked chelate rings. However, in the case of lysine, the ε-amino group can also form a peptide bond with the carboxylic group of another amino acid. This type of peptide amide bond has great importance in biology.3 The ε-(γ-glutamyl)lysine function is one of the covalent crosslink types within and between molecules, and its role is to maintain gross forms of structure and limit the number of degrees of extensibility.4 The amide groups of ε-(glycyl)lysine and ε-(alanyl)lysine form oligopeptide crossbridges in bacterial cell walls, connecting polysaccharide chains, thus forming a polypeptidoglycan network.4,5 The peptide–amide bonds formed with the involvement of the ε-amino groups of lysine or β- and γ-carboxylic groups of aspartic and glutamic acids are separated from the peptide backbone providing new co-ordination environments for metal ions. The present work summarises the results of studies on the copper(II), nickel(II) and zinc(II) complexes of the oligopeptides containing the amide bond on the ε-amino group of lysine, namely Gly-L-Lys(Gly) and α-L-Asp-ε-L-Lys (Scheme 1). | ||
| Scheme 1 | ||
The role of aspartic acid in the binding of metal ions is well known and several papers have already been published concerning transition metal complexes of oligopeptides containing aspartic acid. The data indicate that the effect of the carboxylate groups on the complex formation processes of peptides depends on the location of the aspartyl residue in the peptide chain. The presence of aspartic acid on the N-terminus enhances the metal binding ability of the ligands and slightly suppresses the deprotonation and co-ordination of the amide group as compared to oligoglycines. This effect is especially pronounced if β-carboxylate groups of aspartyl residues are involved in amide binding. The β-carboxylate group at the C-terminus of dipeptides also effects the stability of metal complexes, because its co-ordination results in the enhancement of the stability of the species [ML].6–9
Several papers on the co-ordination chemistry of tri-, tetra- and pentapeptides containing aspartic acid in internal positions have been published; while these peptides can mimic the biologically active sites of the hormone thymopoietin.10–12 It was found that the presence of aspartyl residues increases the metal binding ability of the ligands in all cases and is accompanied by the development of new types of metal ion co-ordination. The aspartyl residue present in the second position of a peptide molecule promotes the metal ion co-ordination of the first amide nitrogen, but significantly suppresses the binding of the subsequent ones. The role of an aspartyl residue present as the third amino acid in a peptide molecule is even more pronounced. The co-ordination of three nitrogen donor atoms (amino and two amides) and the β-carboxylate group of the aspartyl residue is characteristic of all such ligands and tetra- and pentapeptides behave like tripeptides.12,13 Deprotonation and co-ordination of the amide nitrogens takes place cooperatively and the existence of the 2 N-co-ordinated complexes cannot be detected in solution.
The effect of lysyl residues on the thermodynamic stabilities of peptide complexes is much more contradictory. In addition to the aspartyl moieties, the oligopeptide segments of thymopoietin also contain lysyl residues, but they were described as non-co-ordinating side chains in all copper(II) complexes.10–12 Similarly, the potentiometric and spectroscopic data obtained on the copper(II) complexes of GlyHisLys indicated that amino, amide and imidazole nitrogen donor atoms are the major metal binding sites with unco-ordinated and protonated lysyl residues.14–16 On the other hand, the co-ordination of the ε-amino group of lysine was reported to occur in the copper(II) complexes of various dipeptides, resulting in the formation of dinuclear species in solutions,15,17 while one-dimensional polymeric structures were suggested in the solid state.18,19
Copper(II) and nickel(II) complexes of L-γ-Glu-L-ε-Lys and L-Glu-L-ε-Lys were studied by means of potentiometric and spectroscopic techniques.20 In the case of the derivatives of γ-Glu, the amino acid and amide binding sites are too far from each other and the co-ordination of the amide nitrogens is not favoured. The species [ML] predominates in both cases, containing bis(N,O)-bonded metal ions and a fourteen-membered loop around the metal ions. The co-ordination chemistry of L-Glu-L-ε-Lys is reminiscent of common dipeptides, but various polynuclear species are also formed in this case.
These data strongly support that both the co-ordination equilibria and structural variability of the metal complexes of peptides containing ε-lysyl residues are much more complicated than those of common dipeptides, which suggests that further studies are needed in this field.
Experimental
Materials
The peptides GlyLys(Gly) acetate and Asp-ε-Lys were purchased from Bachem and their purities were checked by TLC and HPLC methods. Concentrations of the peptide stock solutions were determined by potentiometric titrations. Stock solutions of copper(II), nickel(II) and zinc(II) chloride were prepared from analytical grade reagents and the concentrations were checked gravimetrically via the precipitation of oxinates.Potentiometric studies
pH potentiometric titrations were performed on 5 cm3 samples in the metal ion concentration range 1.5 × 10−3–8 × 10−3 mol dm−3 with metal ion to ligand ratios between 2 ∶ 1 and 1 ∶ 3. The measurements were made with a Radiometer pHM84 pH meter equipped with a 6.0234.100 combined electrode (Metrohm) and a Dosimat 715 automatic burette (Metrohm) containing a carbonate-free stock solution of potassium hydroxide. 40–60 titration points were recorded for all different ratios. During the titration, argon was bubbled through the samples to ensure the absence of oxygen and carbon dioxide and to stir the solutions. All pH potentiometric measurements were carried out at a constant ionic strength of 0.2 mol dm−3 KCl and at a constant temperature (298 K). The pH readings were converted to hydrogen ion concentration21 and the overall stability constants [log βpqr defined by eqn. (1) and (2)] were calculated by means of a general computational program (PSEQUAD).22| p M + q H + r L ⇌ MpHqLr | (1) |
| βpqr = [MpHqLr]/[M]p[H]q[L]r | (2) |
Spectroscopic studies
UV-VIS absorption spectra of copper(II) and nickel(II) complexes were recorded on a HP 8453 diode array and on a JASCO UVIDEC-610 double-beam spectrophotometer in the same concentration range as used for potentiometry. The absorption maxima and molar absorptivites of various copper(II) complexes were obtained using the PSEQUAD program,22 taking into account the stability constants of complexes and the concentrations and pH values of the individual samples. Anisotropic X-band EPR spectra (9.15 GHz) of copper(II) complexes were recorded at 120 K using a Varian E-9 spectrometer in frozen solutions after addition of ethylene glycol to ensure good glass formation.Results and discussion
Acid–base properties of GlyLys(Gly) and Asp-ε-Lys
The protonation constants of the ligands were determined by pH-metric titrations and the pK values are collected in Table 1. It can be seen from the table that the fully protonated form of GlyLys(Gly) is [H3L]2+ having three dissociable protons in the measurable pH range. Both amino groups are in the vicinity of the amide functions and the corresponding pK values show a close similarity with those of common dipeptides or glycineamide [pK(amino) = 8.13 and 8.01 were reported for diglycine23 and glycineamide,24 respectively]. The average value for the deprotonation of the ammonium groups of GlyLys(Gly) is 8.08 and the difference between them is 0.73 log units, supporting the fact that there is complete overlap in the basicities of the two amino groups. The acidity of the carboxylic function is also in a good agreement with that of diglycine, pK(COOH) = 3.17.23 Asp-ε-L-Lys has four dissociable protons, there being two ammonium and the two carboxylic functions in the molecule. It is important to note that both termini of this peptide molecule can be considered to contain substituted amino acid binding sites. The amino and carboxylic functions of the lysyl residue are well separated from the amide bond and their acid–base properties should be very similar to those of aliphatic α-amino acids.25 As a consequence, the highest (9.66) and lowest (1.91) pK values can be assigned mainly to the amino and carboxylate functions at the lysyl residue. The metal binding sites of the aspartyl residue corresponds to those of β-alanine, but the amino group is very close to the amide function. This results in reduced basicity of the ammonium (pK = 7.79) and reduced acidity of the carboxylic function (pK = 3.11). Of course, the deprotonation of both ammonium and carboxylic groups partly overlap and the exact assignment of the protonation sites is possible only after determination of microscopic protonation constants.| Species | GlyLys(Gly) | Asp-ε-L-Lys |
|---|---|---|
| [HL]− | 8.44(1) | 9.66(1) |
| [H2L] | 16.15(1) | 17.45(2) |
| [H3L]+ | 19.24(2) | 20.56(2) |
| [H4L]2+ | — | 22.47(4) |
| pK1 | 3.09 | 1.91 |
| pK2 | 7.71 | 3.11 |
| pK3 | 8.44 | 7.79 |
| pK4 | — | 9.66 |
Metal complexes of GlyLys(Gly)
The stability constants of the copper(II), nickel(II) and zinc(II) complexes of GlyLys(Gly) were determined by pH potentiometric titrations and the data are collected in Table 2.| Species | Copper(II) | Nickel(II) | Zinc(II) |
|---|---|---|---|
| [MH2L]3+ | 17.96(18) | — | — |
| [MHL]2+ | 14.19(6) | 12.25(5) | 11.42(8) |
| [ML]+ | 9.68(2) | 6.07(2) | 4.74(2) |
| [MH−1L] | 3.54(4) | −3.39(4) | — |
| [ML2] | — | 9.14(8) | 7.00(10) |
| pK([MHL]/[ML]) | 4.51 | 6.18 | 6.68 |
| pK([ML]/[MH−1L]) | 6.14 | 9.46 | — |
| log(K1/K2) | — | 3.00 | 2.48 |
It is clear from Table 2 that the thermodynamic stability of the metal complexes of GlyLys(Gly) follows the Irving–Williams series and formation of bis(ligand) complexes was detected only in the case of nickel(II) and zinc(II). The metal ion speciation of the copper(II)–GlyLys(Gly) system is characterised by the exclusive formation of 1 ∶ 1 complexes at any metal ion to ligand ratio and pH values, as demonstrated by Fig. 1.
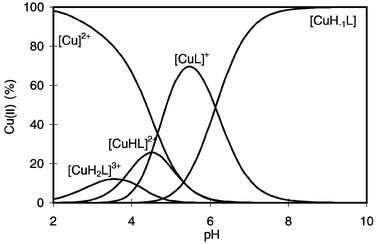 | ||
| Fig. 1 Species distribution of the complexes formed in the copper(II)–GlyLys(Gly) system as a function of pH (cL = cCu = 4 × 10−3 mol dm−3). | ||
It is also clear from Fig. 1 that complex formation in the copper(II)–GlyLys(Gly) system starts in the acidic pH range, demonstrating the high metal binding ability of this peptide molecule. The species [CuH2L]3+ probably has the same co-ordination sites as those of [CuHL]2+, but it is a minor species and independent spectral parameters could not be obtained. The UV-VIS and EPR spectroscopic parameters of the other copper(II) complexes are listed in Table 3 and the corresponding data for GlyGly are also included for comparison.
The EPR parameters in Table 3 suggest that the species [CuH2L]3+ and [CuHL]2+ have the same (NH2,CO) binding sites which are present in both termini of the molecule. Taking into account the similarities in the basicities and metal binding abilities of GlyGly and glycineamide,23,24 any of these residues could be involved in metal binding, while the other donor groups are partially or completely protonated. This means that the species [CuH2L]3+ and [CuHL]2+ have two isomeric forms with the same metal binding sites. As a consequence, neither potentiometric nor visible and EPR spectroscopic techniques can distinguish between these isomers. The formation of [CuL]+ from [CuHL]2+ is accompanied by a significant blue shift of the absorption spectrum suggesting the co-ordination of at least one more nitrogen donor. There are two possibilities for the binding of this nitrogen donor atom; the formation of a bis(NH2,CO) binding site or deprotonation and co-ordination of an amide nitrogen. The formation of a bis(NH2,CO) binding site has already been proposed for the [CuL2] complex of glycylsarcosine, which shows an absorption maximum at 658 nm.23 This value is definitely higher than the absorption maximum of the [CuL]+ complex of GlyLys(Gly), and the steric hindrance also rules out this type of co-ordination in a mononuclear complex. Therefore, the deprotonation and co-ordination of one of the amide nitrogens is more probable, giving a tridentate (NH2,N−,COO−) or bidentate (NH2,N−) binding site at one terminus and a protonated ammonium group at the other. This means that the stoichiometry of this species is [CuH−1LH]+, and the absorption spectra support this assumption. The (NH2,N−,COO−) binding mode was reported to occur in the [CuH−1L] complex of GlyGly, which shows an absorption maximum at 640 nm. Unfortunately, the EPR parameters cannot be compared, because the EPR spectrum of [CuL]+ overlaps with those of other species. The species distribution curves in Fig. 1 partly contradict this observation because the highest concentration of [CuL]+ is 70% of the total metal ion concentration. However, it should be considered that two isomers are possible for the species [CuH−1LH]+. One of the isomers was described above for GlyGly, while the other corresponds to the (NH2,N−) co-ordination of the other terminus (glycineamide-like co-ordination). The EPR parameters of these isomers are only slightly different, resulting in poor resolution of the EPR spectra.
| Species | Co-ordination | λmax/nm; ε/dm3 mol−1 cm−1 | g∥; A∥/10−4 cm−1 |
|---|---|---|---|
| L = GlyLys(Gly) | |||
| [CuH2L]3+ | (NH2,CO) | — | — |
| [CuHL]2+ | (NH2,CO) | 788; — | 2.325; 160 |
| [CuL]+ | (NH2,N−,COO−) | 639; — | — |
| [CuH−1L] | “a”: (NH2,N−,COO−)(NH2,CO) | 623; 89 | “a”: 2.213; 167 |
| “b”: (NH2,CO)(NH2,N−) | “b”: 2.248; 165 | ||
| L = GlyGly23 | |||
| [CuL]+ | (NH2,CO) | — | 2.332; 161 |
| [CuH−1L] | (NH2,N−,COO−) | 640; 84 | 2.248; 185 |
| [CuH−1L2]− | (NH2,N−,COO−)(NH2,CO) | 625, 82 | 2.232; 168 |
It is clear from Fig. 1, and supported by the constancy of the UV-VIS and EPR spectra above pH 7, that the stoichiometry of the major complex is [CuH−1L] and that it is the sole species in neutral and slightly basic solutions. The formation of [CuH−1L] is accompanied by a slight blue shift of the absorption spectrum, which corresponds well to those of [CuH−1L2]− bis(ligand) dipeptide complexes.23 The EPR spectra of equimolar solutions of copper(II) and GlyLys(Gly) at different pH values are shown in Fig. 2 and they suggest a more complex structural behaviour for the species [CuH−1L]. Namely, Fig. 2 shows that two species are in equilibrium with each other above pH 6.6 and the ratio of their concentrations is almost constant at any pH value (approximately 1 ∶ 1). This observation strongly supports the existence of two isomeric forms of [CuH−1L] (see Scheme 2).
 | ||
| Fig. 2 EPR spectra of equimolar solutions of copper(II) and GlyLys(Gly) at different pH values in frozen solution: pH = (a) 4.20, (b) 5.00, (c) 5.81, (d) 6.57, (e) 8.43, (f) 9.64, (g) 10.64, (h) 11.73. | ||
 | ||
| Scheme 2 | ||
The isomer “a” can be described as the tridentate co-ordination of the GlyLys side of the molecule assisted by the bidentate co-ordination of the other glycyl residue to yield a macrochelate with the donor set (NH2,N−,COO−)(NH2,CO). Isomer “b” adopts a bidentate 2N co-ordination of the ε-LysGly side assisted by the bidentate (N,O) co-ordination of the N-terminal Gly residue to yield another macrochelate with the donor set (NH2,N−)(NH2,CO). The composite EPR pattern assigned to the two species was satisfactorily simulated as the sum of two spectra (see Fig. 3). One of them is slightly orthorhombic with A values of 15, 20 and 167 × 10−4 cm−1 and corresponding g values of 2.050, 2.045 and 2.213 (isomer “a” in Fig. 3). The second spectrum is of the axial type with A values of 20, 20 and 165 × 10−4 cm−1 and corresponding g values of 2.060, 2.060 and 2.248 (isomer “b” in Fig. 3). Assigning the spectra is a rather difficult task. However, comparative examination of the EPR parameters measured for a number copper(II) peptide complexes could be of help. Fig. 4 plots g∥versusA∥ values for a series of copper(II) oligopeptide complexes. The open squares represent the mono(chelated) 2N species with (NH2,N−,COO−) co-ordination for dipeptides and (NH2,N−,CO) co-ordination for tri- or tetrapeptides. The solid squares represent the bis(ligand) complexes with bidentate (NH2,CO) co-ordination for the second ligands.
 | ||
| Fig. 3 (a) Experimental EPR spectrum of equimolar solutions of copper(II) and GlyLys(Gly) at pH 9.6 in frozen solution. Spectrum (b) results from the summation of two simulated spectra. (c) Simulated spectra of isomer “a” (dotted line; A values 15, 20 and 167 × 10−4 cm−1, g values 2.050, 2.045 and 2.213) and isomer “b” (solid line; A values 20, 20 and 165 × 10−4 cm−1, g values 2.060, 2.060 and 2.248). | ||
 | ||
| Fig. 4 Plot of g∥ versus A∥ values for a series of copper(II) oligopeptide complexes. The open squares represent the mono(chelated) 2N species with (NH2,N−,COO−) co-ordination for dipeptides and (NH2,N−,CO) co-ordination for tri- or tetrapeptides. The solid squares represent the bis(ligand) complexes with bidentate (NH2,CO) co-ordination for the second ligands. | ||
The arrows in Fig. 4 indicate the changes in the EPR parameters on passing from mono- to bis(ligand) complexes. These changes divide the ligands into two groups. The arrows that correspond to a decrease in g∥ and a small increase in A∥ are characteristic of the complexes of triglycine, tetraglycine and GlyGlyNH2. In these cases, we can suppose that the carbonyl group bound to the metal ion in the equatorial plane in the species [CuH−1L] is replaced by the amino and carbonyl donors of the second ligand molecule. Therefore, the bis(chelated) complex has three nitrogen atoms in the metal plane and a co-ordination geometry that does not deviate significantly from the square planar type. The arrows that show a decrease in both g∥ and A∥ values, a trend that is unusual for a regular geometry, correspond to the formation of bis(ligand) complexes of GlyGly or other common dipeptides. In these cases, we can suppose that the stronger donor carboxylate function cannot be replaced by the second ligand, which will occupy an axial–equatorial position. This results in a distorted structure for the bis(ligand) complex, a situation also described for the [CuH−1L] complex of (GlyCys)2 containing a macrochelate structure with the same binding sites.26 The parameters of isomer “a” of the copper(II)–GlyLys(Gly) system (open triangle in Fig. 4) exhibit a location characteristic of the second class of complexes. Therefore, we can attribute them to isomer “a” having a distorted five-co-ordinate structure. The parameters of isomer “b” have an anomalous position in the plot in Fig. 4 (solid triangle), indicating a different structure. They can be assigned to isomer “b”, but with a distorted co-ordination geometry because of the constraint resulting from the macrochelate. Another important feature of the latter isomer is that it contains only a four-co-ordinate metal ion, the carboxylate function being not involved in metal binding. It should also be noted that the potentiometric measurements could not distinguish between the co-ordination isomers and the differences in the thermodynamic stabilities of the isomers could not be evaluated.
The stability constants of the nickel(II) and zinc(II) complexes of GlyLys(Gly) are also included in Table 2. It is clear from the data that the [NiL]+ and [ZnL]+ complexes are the major species with these metal ions and further deprotonation of these complexes takes place only above pH 9, which is higher than the pK values of the amino groups. As a consequence, the binding sites of the species [ML]+ should be different from those of corresponding copper(II) complexes. The lack of non-co-ordinated and protonated ammonium groups in the species [ML]+ supports the existence of a bis(NH2,CO) chelate, as shown in Scheme 3.
 | ||
| Scheme 3 | ||
The occurrence of similar binding modes has already been reported for the [NiL] species of glycyl-L-cysteine disulfide, containing the bis(NH2,CO) chelate and a macrochelate with the disulfide bridges.26 Of course, the same binding sites should be present in the bis(ligand) complexes of glycineamide, too. The absorption spectra of the [NiL]+ complex of GlyLys(Gly) indicate the presence of an octahedral nickel(II) ion showing 3 absorption maxima at 377, 624 and 980 nm with molar absorptivities of 11, 5 and 6 dm3 mol−1 cm−1, respectively. These values exhibit a very close similarity to those of the nickel(II) complexes of (GlyCys)2 and glycineamide, providing definite proof that the co-ordination geometry is the same.
Further increasing the pH results in precipitation in the zinc(II)-containing system, while the formation of the species [NiH−1L] can be observed with nickel(II). Taking into account the pK values of the free ligand, this reaction should correspond to the deprotonation and co-ordination of one of the amide nitrogen donor atoms, resulting in the same isomeric complexes as reported for copper(II). The pK value for amide deprotonation is higher than those of simple dipeptides,1,2 which can be explained by the extra stabilization caused by the fourteen-membered loop shown in Scheme 3. On the other hand, it also should be noted that the co-ordination geometry of the metal ions is not saturated in the [ML]+ complexes and results in bis(ligand) complex formation with both metal ions. The ratios of the stepwise stability constants (last row in Table 2) are, however, unusually high for octahedral nickel(II) or zinc(II) complexes. These data indicate again the extra stability of the [ML]+ complexes and the minor role of bis(ligand) complex formation.
Metal complexes of Asp-ε-Lys
The stability constants of the metal complexes of Asp-ε-Lys have been determined by potentiometric measurements and the data are summarized in Table 4.| Species | Copper(II) | Nickel(II) | Zinc(II) |
|---|---|---|---|
| [M2L]+ | 14.29(24) | — | — |
| [MHL]+ | 16.44(6) | 14.88(5) | 13.85(5) |
| [M2L2] | 26.74(10) | 18.80(10) | 15.30(7) |
| [M2H−2L2]2− | 7.34(14) | — | — |
| [MH−1L]− | — | — | −2.59(4) |
Before interpreting the data in Table 4, the fact that Asp-ε-Lys is an ambidentate ligand which is capable of several different co-ordination modes should be considered. Namely, the lysyl site of the molecule can be considered as a substituted α-amino acid, while the aspartyl site is a substituted β-alanine with five- and six-membered (NH2,COO−) chelates, respectively. Furthermore, the aspartyl site is considered a substituted amide, having the (NH2,CO) or (NH2,N−) binding sites. As a consequence, the co-ordination equilibria of Asp-ε-Lys are rather complicated, but the model shown in Table 4 gave the best fitting of the potentiometric measurements and is in a good agreement with the spectroscopic parameters. Of course, potentiometric measurements generally cannot differentiate between mononuclear and polynuclear species with the same stoichiometries, thus the involvement of the species [CuL] and [CuH−1L]− instead of [Cu2L2] and [Cu2H−2L2]2− gave almost the same fitting, but they contradict the spectroscopic measurements.
The species distribution of the complexes formed in the copper(II)–Asp-ε-Lys system is reported in Fig. 5 and shows the exclusive formation of the dinuclear species [Cu2L2] and [Cu2H−2L2]2− in neutral and basic solution.
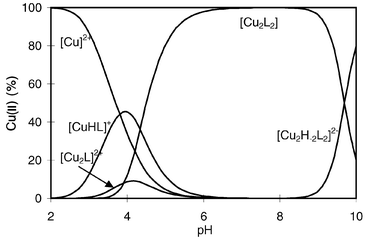 | ||
| Fig. 5 Species distribution of the complexes formed in the copper(II)–Asp-ε-Lys system as a function of pH (cL = cCu = 4 × 10−3 mol dm−3). | ||
The species [CuHL]+ was detected in acidic media and there are several possibilities for its metal ion co-ordination. It can be seen from Fig. 5 that pH 4 is the best condition for the formation of [CuHL]+, and comparison of the literature data on the metal complexes of α- and β-amino acids23,25,27 reveals that glycine-like (NH2,COO−) or glycineamide-like (NH2,CO) five-membered chelates are most favoured under these conditions. The high pK values and low metal binding affinity of the β-alanine sites of the aspartyl residue shift the complex formation processes of β-alanine to higher pH values. Taking into account the higher thermodynamic stability of the metal complexes of α-amino acids compared to those of the corresponding amides, the α-amino acid-like (NH2,COO−) co-ordination should be preferred. The EPR parameters of the complex [CuHL]+ are, however, g∥ = 2.304 and A∥ = 164 × 10−4 cm−1 and these values are distinctly different from the parameters of [CuL]+ formed with either α-alanine (g∥ = 2.300 and A∥ = 177 × 10−4 cm−1) or glycineamide (g∥ = 2.331 and A∥ = 161 × 10−4 cm−1) supporting the existence of an equilibrium between the two co-ordination modes.
The major species is formed at pH 6 and its stoichiometry can be given as [CuL] or [Cu2L2]. Steric requirements rule out the co-ordination of both termini to the same metal ion, therefore the mononuclear complex [CuL] should be a co-ordinatively unsaturated complex. The presence of such a species would result in bis(ligand) complex formation, but this was not detected at any pH values and metal ion to ligand ratios. The spectroscopic parameters provide further support for dinuclear complex formation. The significant line broadening of the spectra (see Fig. 6) and the detection of ΔM = 2 resonances indicate that the dinuclear model is more reliable. In particular, [Cu2L2] yields a broad resonance at gca. 2, whereas [Cu2H−2L2]2− is characterised by a powder-like spectrum, indicative mainly of a dipolar-type interaction between the metal ions. The ΔM = 2 resonances were detected for both species. The suggested co-ordination modes of the species [Cu2L2] and [Cu2H−2L2]2− are shown in Scheme 4 and 5, respectively.
 | ||
| Fig. 6 EPR spectra of equimolar solutions of copper(II) and Asp-ε-Lys at different pH values in frozen solution: pH = (a) 6.16, (b) 7.45, (c) 9.22, (d) 10.26, (e) 11.09. | ||
 | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
According to Scheme 4 the species [Cu2L2] is the best described as a dinuclear complex with amino acid binding sites. The d–d absorption maximum (λmax = 644 nm) is in a good agreement with the co-ordination of 2N donors. In the case of α-alanine or other α-amino acids, the absorption maxima occur at slightly lower wavelengths (620–630 nm), but in this case, both five-and six-membered chelates are involved in the metal binding. In principle, there are two possibilities for the formation of the dinuclear complex: (i) containing mixed five-and six-membered chelates (like Scheme 4), or (ii) containing separated bis(5) and bis(6)-membered chelates. Previous results on the mixed ligand complexes indicated that the order of stability of copper(II) mixed ligand complexes follows the trend: (5,6) ≥ (5,5) > (6,6).28–30 This means that bis(chelate) complexes comprising two six-membered chelates are much less stable than the other systems. As a consequence, the dinuclear species containing separated bis(5) and bis(6)-membered chelates would not be stable enough for the exclusive formation of the species [Cu2L2].
A new base-consuming process starts above pH 9 and is accompanied by a significant blue shift of the absorption maximum (λmax = 585 nm), suggesting the co-ordination of one more nitrogen donor atom. EPR spectra definitely prove the existence of a dinuclear species [Cu2H−2L2]2− and its binding sites are best represented by Scheme 5. In other words, deprotonation and co-ordination of the amide nitrogen result in the replacement of the six-membered (NH2,COO−) chelate with the five-membered (NH2,N−) chelate, which is further stabilized by the axial co-ordination of the carboxylate residue. As a result, the dinuclear species again adopts a symmetrical arrangement consisting of two metal ions and two ligands connected by peptide chains.
Comparison of the data in Table 4 reveals that the formation processes of nickel(II) and zinc(II) complexes with Asp-ε-Lys are slightly different from those of copper(II). Of course, potentiometric data do not provide final proof of the structures of the various species, but the binding sites of the ligands can be evaluated by the comparison of literature data. Namely, the formation of the species [MHL]+ is probably best described by the co-ordination of the amino and carboxylate donor groups of the lysyl or aspartyl residues in the form of five- or six-membered chelates, while the other amino groups remain protonated. Taking into account the pK values of these amino groups, logK = 7.09 and 5.22 can be obtained for the stability of the five-and six-membered (NH2,COO−)-co-ordinated complexes of nickel(II), respectively. These values should be compared to those of [NiL]+ with α-alanine (logK1 = 5.41) and β-alanine (logK1 = 4.55). In the case of zinc(II), similar treatment of the equilibrium data provides the values logK = 6.06 and 4.19 for Asp-ε-Lys, which should be compared with those for the [ZnL]+ complexes of α-alanine (logK1 = 4.63) and β-alanine (logK1 = 4.14).25 It is evident from the previous data that the formation of [NiHL]+ and [ZnHL]+ complexes shows some extra stabilization as compared to the common amino acid binding modes. At the same time, the parameters obtained for copper(II) complexes of Asp-ε-Lys are logK = 8.65 and 6.78, which are in reasonable agreement with the parameters of the [CuL]+ complexes of α-alanine (logK1 = 8.14) and β-alanine (logK1 = 6.99).25 The co-ordination geometry of the copper(II) complexes is, however, best described by a distorted octahedron containing bidentate amino acid-like co-ordination in the [CuHL]+ species of Asp-ε-Lys. On the other hand, the regular octahedral geometry of nickel(II) and zinc(II) prefers the tridentate co-ordination of Asp-ε-Lys in the corresponding [MHL]+ complexes, containing the (NH2,COO−) co-ordinations, assisted by the metal binding of another carboxylate or carbonyl residue.
Computer evaluation of the potentiometric data provides reasonable fitting with the involvement of either mononuclear [ML] or dinuclear complexes [M2L2] in the model at higher pH values. On the basis of pure potentiometric data, we cannot distinguish between these models, but it should be considered that formation of bis(ligand) complexes was rejected in any computational models, similarly to copper(II) complexes. Steric requirements, however, rule out the saturation of the co-ordination sphere of the metal ions in the mononuclear complexes, providing indirect proof of the existence of dinuclear species. The structure of the dinuclear species is best described by the similar amino acid binding sites, as shown for the corresponding copper(II) complexes in Scheme 4. The major difference in the complex formation reactions of copper(II) and the other two metal ions is reflected in the absence of amide deprotonation and co-ordination with nickel(II) and zinc(II). In the case of zinc(II) complexes, this is not surprising, because zinc(II) ions are generally not able to induce co-ordination of the peptide amide bonds to a metal ion, except for some specific amino acid sequences. The species [ZnH−1L]− formed above pH 8 is a mixed hydroxo complex and is followed by precipitation of hydroxides at higher pH values. However, nickel(II) forms stable complexes with both di-and tripeptides in slightly basic solution.1,2 The formation of stable chelate complexes generally suppresses these deprotonation reactions, as was reported for the nickel(II) complexes of AspPheNH2.31 In the case of glycyl-L-cysteine disulfide, the bis(chelate) binding is supported by the formation of a loop around nickel(II) and this results in extra stability without amide binding.26 The complex formation reactions of the nickel(II)–Asp–ε-Lys system can probably be explained in a similar way: the extra stabilization of the dinuclear complex hinders co-ordination of the amide functions to the metal ion.
Conclusions
The results obtained on the transition metal complexes of peptides containing the ε-amino groups of lysyl residues in the amide bond, GlyLys(Gly) and Asp-ε-Lys, reveal great variety in the complex formation processes of these ligands as compared to common di- or tripeptides. Both peptides can be considered as ambidentate ligands, and their co-ordination modes are a function of the nature of the metal ions and the pH of the solutions.GlyLys(Gly) is built up from 3 amino acids, but its co-ordination chemistry is completely different from those of common tripeptides. It can co-ordinate to the metal ions via the amino and carbonyl groups at both termini or, after deprotonation of the amide functions, it can be considered either as a substituted dipeptide or a substituted glycineamide. The stoichiometry of the major species is [CuH−1L] in the copper(II)–GlyLys(Gly) system and EPR spectroscopy unambiguously proves the existence of two isomeric forms of the complex. The extra stability of these isomers arises from the formation of chelate rings coupled via a macrochelate [Scheme 2(a), (b)] and suppresses hydrolytic reactions. The nickel(II)– and zinc(II)–GlyLys(Gly) systems are characterised by the formation of stable [ML]+ complexes and the increased stability of these species is explained by bis(NH2,CO) co-ordination coupled via a macrochelate (see Scheme 3). Deprotonation and co-ordination of the amide nitrogens were, however, detected only in the copper(II) and nickel(II) complexes of GlyLys(Gly).
The co-ordination chemistry of Asp-ε-Lys is best characterised by the presence of amino acid binding sites, similar to those of α- and β-alanine. In addition, it can behave as a substituted glycineamide with a very high affinity for dimerisation. The stoichiometries of the dinuclear complexes can be given as [M2L2] and [M2H−2L2]2−, containing the binding modes shown in Scheme 4 and 5. In the case of copper(II), the formation of both dinuclear species has been detected, in agreement with the high affinity of copper(II) for binding to amide nitrogens of peptides. However, in the nickel(II)– and zinc(II)–Asp-ε-Lys systems, only the species [M2L2] was detected, containing the amino acid binding sites and suppressing deprotonation of both amide groups and co-ordinated water molecules.
Acknowledgements
This work was supported by the Hungarian National Research Fund (OTKA T 034361).References
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385 CrossRef CAS.
- I. Sóvágó, in Biocoordination Chemistry, ed. K. Burger, Ellis Horwood, New York, 1990, pp. 135–184 Search PubMed.
- J. E. Folk and J. S. Finlayson, Adv. Protein Chem., 1977, 31, 1 Search PubMed.
- J. L. Strominger and J.-M. Ghuysen, Science, 1967, 156, 213 CAS.
- J.-M. Ghuysen, J. L. Strominger and D. J. Tipper, Comp. Biochem. Physiol., B: Biochem. Mol. Biol., 1968, 26, 53 Search PubMed.
- A. Gergely and E. Farkas, J. Chem. Soc., Dalton Trans., 1982, 381 RSC.
- B. Decock Le-Reverand, F. Liman, C. Livera, L. D. Pettit, S. I. Pyburn and H. Kozlowski, J. Chem. Soc., Dalton Trans., 1988, 887 RSC.
- J.-F. Galey, B. Decock Le-Reverand, A. Lebkiri, L. D. Pettit, S. I. Pyburn and H. Kozlowski, J. Chem. Soc., Dalton Trans., 1991, 2281 RSC.
- I. Sóvágó, E. Farkas, C. Bertalan, A. Lebkiri, T. Kowalik-Jankowska and H. Kozlowski, J. Inorg. Biochem., 1993, 51, 715 CrossRef CAS.
- I. Sóvágó, T. Kiss and A. Gergely, Inorg. Chim. Acta, 1984, 93, L53 CrossRef CAS.
- I. Sóvágó, B. Radomska, I. Schön and O. Nyéki, Polyhedron, 1990, 9, 825 CrossRef CAS.
- I. Sóvágó, C. Bertalan, L. Göbl, I. Schön and O. Nyéki, J. Inorg. Biochem., 1994, 55, 67 CrossRef CAS.
- B. Decock Le-Reverand, A. Lebkiri, C. Livera and L. D. Pettit, Inorg. Chim. Acta, 1986, 124, L19 CrossRef.
- P. May, J. Whittaker and D. Williams, Inorg. Chim. Acta, 1983, 80, 5 CrossRef CAS.
- M. J. A. Rainer and B. M. Rode, Inorg. Chim. Acta, 1985, 107, 127 CrossRef CAS.
- H. Kozlowski, W. Bal, M. Dyba and T. Kowalik-Jankowska, Coord. Chem. Rev., 1999, 184, 319 CrossRef CAS.
- B. Radomska, I. Sóvágó and T. Kiss, J. Chem. Soc., Dalton Trans., 1990, 289 RSC.
- B. Radomska, M. Kubiak, T. Glowiak, H. Kozlowski and T. Kiss, Inorg. Chim. Acta, 1989, 159, 111 CrossRef CAS.
- G. Fusch, E. C. Hillgeris and B. Lippert, Inorg. Chim. Acta, 1994, 217, 33 CrossRef CAS.
- P. G. Daniele, E. Prenesti and G. Ostacoli, J. Inorg. Biochem., 1996, 61, 165 CrossRef CAS.
- H. Irving, G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475 CrossRef CAS.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Stability Constants, ed. D. Leggett, Plenum Press, New York, 1985, p. 291 Search PubMed.
- I. Sóvágó, D. Sanna, A. Dessi, K. Várnagy and G. Micera, J. Inorg. Biochem., 1996, 63, 99 CrossRef CAS.
- I. Sóvágó, B. Harman, A. Gergely and B. Radomska, J. Chem. Soc., Dalton Trans., 1986, 235 RSC.
- I. Sóvágó, T. Kiss and A. Gergely, Pure Appl. Chem., 1993, 65, 1029 CAS.
- Cs. G. Ágoston, K. Várnagy, A. Bényei, D. Sanna, G. Micera and I. Sóvágó, Polyhedron, 2000, 19, 1849 CrossRef CAS.
- T. Kiss, I. Sóvágó and A. Gergely, Pure Appl. Chem., 1991, 63, 597.
- H. Sigel, R. Caraco and B. Prijs, Inorg. Chem., 1974, 13, 462 CrossRef CAS.
- I. Sóvágó and A. Gergely, Inorg. Chim. Acta, 1979, 37, 233 CrossRef CAS.
- H. Sigel, B. Prijs and R. B. Martin, Inorg. Chim. Acta, 1981, 56, 45 CrossRef CAS.
- K. Várnagy, H. Kozlowski, I. Sóvágó, T. Kowalik-Jankowska, M. Kruszynski and J. Zboinska, J. Inorg. Biochem., 1988, 34, 83 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |