[Triangulo-Hg3(μ-dppm)3](O3SCF3)4: first complete NMR analysis of a homoleptic [M3(μ-dppm)3] type system and study of the conformation dynamics in solution by use of its dpam/mdppm derivatives
Walter
Schuh
a,
Gerhard
Hägele
*b,
Ralf
Olschner
b,
Andreas
Lindner
b,
Peter
Dvortsak
c,
Holger
Kopacka
a,
Klaus
Wurst
a and
Paul
Peringer
*a
aInstitut für Allgemeine, Anorganische und Theoretische Chemie, Universität Innsbruck, Innrain 52a, A-6020 Innsbruck, Austria. E-mail: paul.peringer@uibk.ac.at
bInstitut für Anorganische und Strukturchemie, Heinrich Heine Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: haegele@uni-duesseldorf.de
cBruker Analytik GmbH, Silberstreifen, D-76287 Rheinstetten, Germany
First published on 27th November 2001
Abstract
The 31P and 199Hg spectra of the new subvalent [triangulo-Hg3]4+ based clusters [Hg3(μ-mdppm)3](O3SCF3)4 (1a; mdppm = Ph2PCH(Me)PPh2), which is formed stereoselectively with a syn–anti–anti orientation of the methyl groups, and [Hg3(μ-dppa)3](O3SCF3)4 (4; dppa = Ph2PNHPPh2) are successfully simulated with the program WIN-DAISY. By use of the parameters derived from 1a and 4 the spectra of the parent system [Hg3(μ-dppm)3](O3SCF3)4, which show a very complex pattern, are for the first time completely analysed. The simplification of the spectral pattern in 1a or 4 compared to [Hg3(μ-dppm)3](O3SCF3)4, respectively, is a result of either symmetry reduction (1a) or quasi selective variation of one Hg–P coupling constant (4). For δ(31P), δ(199Hg), J(HgHg) and the different classes of P–P and Hg–P couplings specific values are found, some of which are powerful tools for the identification of [triangulo-Hg3]4+ compounds. The 1H NMR parameters of 1a, anti- and syn-[Hg3(μ-dpam)(μ-mdppm)2](O3SCF3)4 (2a, 2b; dpam = Ph2AsCH2AsPh2), [Hg3(μ-dpam)2(μ-mdppm)](O3SCF3)4 (3) and [Hg3(μ-dpam)3](O3SCF3)4 lead to a rational interpretation of conformation dynamics in these systems: The three five-membered rings formed by the edges of the [triangulo-Hg3]4+ cluster and the three bridging ligands adopt C-envelope conformations in a way that two of the flaps are oriented above and one below the Hg3 plane, and vice versa. The mdppm ligands feature a rigid conformation with an equatorial methyl group and an axial hydrogen atom at the flap carbon atom. The methylene carbon atom of the dpam ligands is able to flip between two positions above and below the Hg3 plane, if the relative orientations of the three flaps mentioned above are retained. The overall found rigid conformation of the bridging mdppm ligand allows a qualitative estimation of relative thermodynamic preferences of isomers resulting from syn or anti orientation of mdppm methyl groups.
Introduction
Many compounds containing a [M3(μ-LL)3] subunit, where M is Ni,1 Pd,2 Pt,3 Cu,4 Ag,5 Au6 or Hg7 and LL is dppm or a related bidendate ligand,8 have been synthesised during the last three decades. These complexes have attracted considerable attention due to their role as anion recognition hosts,2c,d,4,5,7c,e in view of their photophysical properties2b,4a,b,9 and because of their catalytic activity.1,3,10 Whereas these compounds are well characterised in the solid state by single crystal X-ray diffraction, the reported solution NMR data are restricted to 1H, 31P, 195Pt and 199Hg chemical shifts as well as some coupling constants obtained from partial analysis of the spectra or from heteronuclear clusters.11 Difficulties in analysing, in particular, 31P{1H} NMR spectra arise from their complex pattern in the case of compounds involving metal isotopes with I = ½ or from spectra consisting of a single resonance line for compounds containing metal isotopes with I ≠ ½. Multinuclear NMR spectroscopy is apparently the most important tool for the structural characterisation of compounds in solution, revealing information about atomic connectivities, molecular conformation, intra- and inter-molecular exchange processes. As the first example for [M3(μ-dppm)3] type systems we will show for [Hg3(μ-dppm)3](O3SCF3)4, that consequent derivatisation techniques combined with extensive NMR spectroscopic investigations lead to a complete set of 1H, 31P and 199Hg NMR shifts and coupling constants, which allow an unambiguous identification of such compounds in solution and give insights into their conformation dynamics. The identity of the cluster [Hg3(μ-dppm)3]4+ with an average formal oxidation state of +4/3 for mercury has been established by single crystal X-ray diffraction.7a,c Phosphorus-31 and mercury-199 NMR spectroscopy is expected to yield interesting parameters but the analysis of the complex spectra was hitherto unsuccessful. Fig. 1 shows the 31P{1H} NMR spectrum of [Hg3(μ-dppm)3](O3SCF3)4. The satellite pattern due to isotopomers containing one 199Hg nucleus consists of a multitude of transitions extended over a range of ca. 2000 Hz and emerges from the baseline only by drastic vertical expansion.4 (center at 54.8 ppm, spectral width 2300 Hz).](/image/article/2002/DT/b102840p/b102840p-f1.gif) | ||
| Fig. 1 31P{1H} NMR spectrum of [Hg3(μ-dppm)3](O3SCF3)4 (center at 54.8 ppm, spectral width 2300 Hz). | ||
First insights into 31P and 199Hg NMR parameters of [Hg3]4+ clusters were gained recently by derivatisation of [Hg3(μ-dppm)3](O3SCF3)4: Systematic substitution of dppm by dpam and Ph2PCH2AsPh2 resulted in new compounds with lower P/[Hg3]4+ ratio and reduced symmetry, which enabled a successful analysis of their NMR spectra.7d
In this article we report on the synthesis of [Hg3(μ-mdppm)3](O3SCF3)4 (1a; Scheme 1) and [Hg3(μ-dppa)3](O3SCF3)4 (4; Scheme 1). The NMR spectra of both clusters are easier to analyse compared with [Hg3(μ-dppm)3](O3SCF3)4 because of the inequivalence of the phosphorus atoms in the homoleptic complex 1a (see below) and a favourable constellation of the Hg–P coupling constants of 4. Using the data of 1a and 4 enables a successful interpretation of the 31P and 199Hg NMR patterns of [Hg3(μ-dppm)3](O3SCF3)4. Furthermore 1H NMR spectroscopy of the new dpam/mdppm mixed ligand clusters anti- and syn-[Hg3(μ-dpam)(μ-mdppm)2](O3SCF3)4 (2a, 2b; Scheme 1) and [Hg3(μ-dpam)2(μ-mdppm)](O3SCF3)4 (3; Scheme 1) together with the homoleptic clusters 1a and [Hg3(μ-dpam)3](O3SCF3)4 reveals detailed information about conformation dynamics of these compounds in solution.
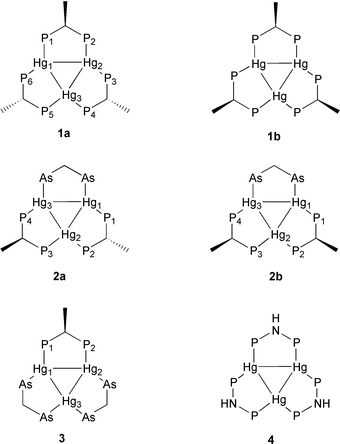 | ||
| Scheme 1 Structures of the compounds 1a, 1b, 2a, 2b, 3 and 4. The charge (4+), phenyl rings and O3SCF3− anions are omitted for clarity. | ||
Results and discussion
Syntheses and stereochemistry
| 2 [Hg(Me2SO)6](O3SCF3)2 + Hg + 3 mdppm → 1a + 12 Me2SO | (1) |
| 2 Hg2(O3SCF3)2 + 3 mdppm → 1a + Hg | (2) |
The 31P{1H} NMR spectrum of the isotopomer without 199Hg nuclei (see NMR section) consists of an [ABM]2 pattern (Fig. 2) in contrast to [Hg3(μ-dppm)3](O3SCF3)4 for which a [[A]2]3 spin system gives rise to a single resonance line. The inequivalence of the phosphorus atoms of 1a is attributed to the relative orientation of the methyl groups of the mdppm ligands, consistent with a syn–anti–anti arrangement as shown in Scheme 1. Interestingly, the isomer containing all methyl groups in a syn orientation (1b, Scheme 1) is not formed at all. We suppose that the exclusive formation of the isomer 1a is due to the thermodynamic preference of an “all-equatorial” orientation of the methyl groups of the mdppm ligands: All crystal structures of [Hg3(μ-dppm)3]4+ clusters exhibit envelope conformations for the three Hg2P2C five-membered rings with the carbon atom at the flap, two of the flaps are positioned above and one below the Hg3 plane as sketched in Scheme 2.7a,c Each flap bears an equatorial and an axial methylene proton. We propose similar relationships for 1a and that the equatorial positions are occupied by methyl groups of the mdppm ligands. There is NMR spectroscopic evidence that this also applies for 2a, 2b and 3 (see below). For the syn–syn–syn isomer 1b there is no opportunity for an “all-equatorial” arrangement of the methyl groups with a simultaneous C-envelope conformation of the Hg2P2C rings and two of the flaps oriented above and one below the Hg3 plane.
![Conformation of [Hg3(μ-dppm)3]4+ complexes in the solid state (schematic representation); a and e denote axial and equatorial positions at the envelope carbon atoms.](/image/article/2002/DT/b102840p/b102840p-s2.gif) | ||
| Scheme 2 Conformation of [Hg3(μ-dppm)3]4+ complexes in the solid state (schematic representation); a and e denote axial and equatorial positions at the envelope carbon atoms. | ||
 | ||
| Fig. 2 Experimental (upper trace) and simulated (lower trace) 31P{1H} NMR spectrum of 1a (center at 59.8 ppm, spectral width 3500 Hz). | ||
There is another example of a significant thermodynamic preference of one diastereomer in a Hg/mdppm system reported in the literature: Dean and Srivastava observed the presence of two isomers of [Hg2(μ-mdppm)2]4+ in which two Hg2+ nuclei are bridged by mdppm ligands.12 The occurrence of isomerism was attributed to syn and anti arrangements of the methyl groups. An assignment of the 31P signals to syn and anti isomers could not be made. Equilibrated solutions of [Hg2(μ-mdppm)2]4+ show a ratio [major isomer]/[minor isomer] > 10.
Two other binuclear, doubly bridged metal complexes of mdppm have been prepared: 1H and 31P{1H} NMR data of [Pd2(μ-mdppm)2Cl2] revealed the presence of two isomers in a molar ratio of 2/1, of which the major product has been shown by single crystal X-ray diffraction to display an anti arrangement of the methyl groups.13 For [Ag2(μ-mdppm)2](BF4)231P{1H} solution NMR data indicate the formation of a sole diastereomer, unfortunately no X-ray crystal structure determination has been performed to establish the relative orientations of the methyl groups.14
| 2 [Hg(Me2SO)6](O3SCF3)2 + Hg + dpam + 2 mdppm → 2a, 2b + 12 Me2SO | (3) |
The clusters 2a with the methyl groups in anti arrangement and 2b with the methyl groups in syn arrangement are present in a ratio of 2/1. According to 1H NMR results (see below) we presume two energetically degenerate orientations of the methylene group of the dpam ligand in 2a, whereas only one orientation is present in 2b. We think that this causes the thermodynamic preference of the diastereomer 2a.
Compound 3 is formed according to eqn. (4). The clusters 2a and 2b (together ca. 20%) and [Hg3(μ-dpam)3](O3SCF3)4 (ca. 20%) were identified as byproducts.
| 2 [Hg(Me2SO)6](O3SCF3)2 + Hg + 2 dpam + mdppm → 3 + 12 Me2SO | (4) |
The clusters 2a, 2b and 3 could not be obtained analytically pure in the solid state and were identified by 31P{1H}, 199Hg{1H} and 1H NMR spectroscopy.
1H NMR conformational studies of 1a, 2a, 2b, 3 and [Hg3(μ-dpam)3](O3SCF3)4
The compounds 1a, 2a, 2b, 3 and [Hg3(μ-dpam)3](O3SCF3)4 with the ligands mdppm and dpam in various stoichiometries exhibit distinctly separated chemical shift ranges for the different aliphatic hydrogen atoms: δ(CH)mdppm 4.43–4.24, δ(CH2)dpam 3.75–3.19, δ(CH3)mdppm 1.70–1.50. The H–H and P–H coupling constants do not show any peculiarities, but there are some remarkable features concerning the CH2-hydrogen atoms of the dpam ligand: Compared with the methine and methyl hydrogen atoms of the mdppm ligand, the methylene protons of dpam show a relatively wide distribution of their chemical shift values. Furthermore the 3J(HgH) coupling constants for the CH2-hydrogens in dpam involve values between 0 and 56 Hz whereas for the aliphatic CH-hydrogens of mdppm this coupling constant is in all cases too small to be detected. Two assumptions are made for the subsequent discussion to allow a reasonable interpretation for these observations:1. The Hg2P2C and Hg2As2C five-membered rings adopt an envelope conformation with the carbon at the flap even in solution (based on crystallographic data of various [Hg3(μ-dppm)3]4+ complexes)7a,c and
2. The 3J(Hg–P–C–H)/3J(Hg–As–C–H) coupling constants show a dihedral angular dependence analogous to the Karplus–Conroy relationship for vicinal 3J(H–C–C–H) coupling constants (based on the fact that a similar dependence has been observed for 3J(Pt–P–C–H) couplings in platinum complexes containing dppm as ligand).15
Consequently the value of 3J(Hg–P–C–H)/3J(Hg–As–C–H) is expected to be approximately 0 Hz for a hydrogen atom in an axial position (ϕca. 90°), whereas a hydrogen atom in an equatorial position (ϕca. 180°) should exhibit a markedly higher value.
No 3J(Hg–P–C–H) coupling has been observed for the aliphatic methine protons of the mdppm ligand in the compounds 1a and 3. This indicates an axial position for these hydrogen atoms. Consequently compound 1a adopts a conformation with two carbon atoms of the three five membered rings positioned below and one above the Hg3-plane. The mdppm CH-signals of the compounds 2a and 2b could not be analysed due to violent line overlapping but the hydrogen atoms are thought to adopt an axial position as well: This is indicated by their small chemical shift difference compared with the methine protons in compounds 1a and 3 (<0.2 ppm), whereas the axial and equatorial hydrogen atoms of the dpam CH2-group in compound 2b show a chemical shift difference of more than 0.5 ppm (see below).
In contrast to dppm in the complex [Hg3(μ-dppm)3]4+ and dpam in [Hg3(μ-dpam)3]4+ (see below) the mdppm ligand features a rigid structure in solution, the Hg2P2C five-membered ring adopts a C-envelope conformation with an axial methine hydrogen and an equatorial methyl group. A similar conformation has been found in the crystal structure of anti-[Pd2(μ-mdppm)2Cl2] for the Pd2P2C five-membered rings.13b
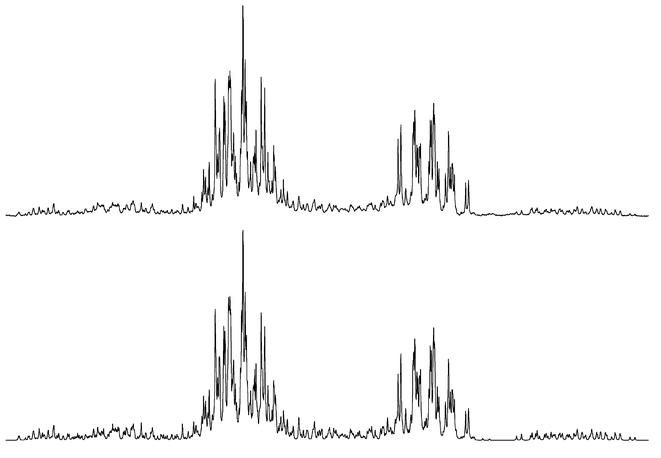
The methylene protons of the dpam ligand in compound 2b represent an AB system with a chemical shift difference of ΔδAB = 0.56 ppm and two 3J(Hg–As–C–H) coupling constants having values of 0 and 56 Hz. These observations suggest a rigid conformation of the Hg2As2C five-membered ring with fixed positions of the methylene hydrogen atoms. It is most probable that the methylene carbon atom of the dpam ligand in 2b points to the other side of the Hg3 plane relative to the methine carbon atoms of the mdppm ligands. The assignment of Ha and Hb is based on this consideration (Scheme 3).
 | ||
| Scheme 3 Assignment of Ha and Hb at the dpam methylene carbon atoms in compounds 2b and 3 according to Table 3. | ||
As found for the chemical shifts of the axial and equatorial hydrogen atoms in cyclohexane (chair conformation),16 the axial dpam methylene proton in compound 2b shows its resonance shifted to lower frequencies with respect to the equatorial proton.
In compound 2a the chemical shift of the dpam methylene protons and the Hg–As–C–H coupling constant show values of 3.41 ppm and 27 Hz, which approximately coincide with the mean values of δ(Ha)/δ(Hb) and 3J(HgHa)/3J(HgHb) in 2b (3.47 ppm, 28 Hz). This observation is interpreted in terms of a rapid interconversion of the two C-envelope conformations of the Hg2As2C five-membered ring, where the methylene carbon atom is placed either below or above the Hg3 plane. Taking into account the rigid structure for the Hg2P2C five-membered rings involving the mdppm ligands, compound 2a consecutively exhibits a structure with two carbon atoms of the three five-membered rings positioned below and one above the Hg3-plane.
The dpam methylene protons in compound 3 show Hg–H coupling constants of 16 and 43 Hz. Because of the fixed conformation of the mdppm ligand with the methyl group in an equatorial orientation, there are three possibilities for the two dpam ligands to conform with the structure mentioned above: Both dpam methylene carbon atoms point to the opposite side of the Hg3-plane relative to the mdppm methine carbon atom (A), or one dpam methylene carbon atom is below and the other above the Hg3-plane, and vice versa (B and C). There are Hg–H couplings of ca. 60 Hz and ca. 0 Hz expected for conformer A. The structures B and C are anticipated to exhibit a ca. 30 Hz coupling for both protons provided for rapid interconversion of B and C. The actual observation of coupling constants of 16 and 43 Hz thus indicates the simultaneous presence of A, B and C, a slightly lower energy content of A compared with B and C and a rapid interconversion of A, B and C on the 1H NMR time scale. The hydrogen atoms exhibiting a higher axial probability are assigned to Ha (smaller value of 3J(Hg–As–C–H)), the hydrogen atoms adopting more often an equatorial position are assigned to Hb (higher value of 3J(Hg–As–C–H)) as indicated in Scheme 3.
The compound [Hg3(μ-dpam)3](O3SCF3)4, present as byproduct in solutions of 3, features a 3J(Hg–As–C–H) of 32 Hz. By comparison to the related coupling constants in the compounds 2a, 2b and 3 this value leads to the proposal of a rapid interconversion of the six possible conformers (Scheme 4). The apparent value of 3J(Hg–As–C–H) is interpreted as the mean of the couplings between mercury and the axial and the equatorial hydrogen, respectively. The same dynamic behaviour proposed for [Hg3(μ-dpam)3](O3SCF3)4 is suggested for [Hg3(μ-dppm)3](O3SCF3)4 (3J(Hg–P–C–H) = 41 Hz) as well.
4 in solution (schematic representation).](/image/article/2002/DT/b102840p/b102840p-s4.gif) | ||
| Scheme 4 Proposed conformation dynamics for [Hg3(μ-dpam)3](O3SCF3)4 in solution (schematic representation). | ||
The 1H NMR parameters of the compounds 1a, 2a, 2b, 3 and [Hg3(μ-dpam)3](O3SCF3)4 are collected in Table 1.
| 1a | 2a | 2b b | 3 b | [Hg3(μ-dpam)3](O3SCF3)4 | ||||
|---|---|---|---|---|---|---|---|---|
| a Standard deviations for chemical shifts and coupling constants calculated by the WIN-DAISY automatic routine are ≤0.5 Hz. b For assignment of Ha and Hb see Scheme 3. | ||||||||
| δ(CH)mdppm | 4.24 (2H) | 4.36 (1H) | 4.43–4.24 | 4.43–4.24 | 4.43 | — | ||
| 3 J(CH–CH3)/Hz | 7.6 | 6.7 | 7.4 | — | ||||
| 2 J(P–CH)/Hz | 13.4, 13.4 | 14.1 | 15.0 | — | ||||
| δ(CH3)mdppm | 1.50 (6H) | 1.63 (3H) | 1.7–1.5 | 1.7–1.5 | 1.70 | — | ||
| 3 J(P–CH3)/Hz | 11.4, 11.3 | 9.8 | 11.7 | — | ||||
| δ(CH2)dpam | — | 3.41 | 3.19 (Ha) | 3.75(Hb) | 3.47(Ha) | 3.72(Hb) | 3.72 | |
| 2 J(H–C–H)/Hz | — | −11.8 | −11.8 | −11.9 | −11.9 | |||
| 3 J(Hg–CH2)/Hz | — | 27 | ≈0 | 56 | 16 | 43 | 32 | |
| Solvent | CD2Cl2–MeOH (2/1) | CD2Cl2 | CD2Cl2 | CD2Cl2 | CD2Cl2 | |||
Analysis of the 31P and 199Hg NMR spectra
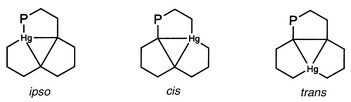
The P–P and Hg–P coupling constants are denoted according to Schemes 5 and 6, respectively.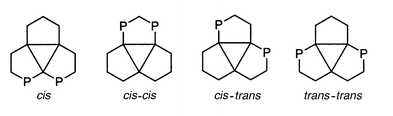 | ||
| Scheme 5 Stereochemical relations between two P atoms (schematic representation); relations are designated according to the shortest coupling pathway along the Hg3 framework. | ||
 | ||
| Scheme 6 Stereochemical relations between mercury and phosphorus (schematic representation); relations are designated according to the shortest coupling pathway along the Hg3 framework, J(HgP)ipso is always denoted as 1J(HgP) in the text. | ||
All simulations and iterations were performed by use of the program WIN-DAISY.17 During the last iteration cycle all spectral parameters (chemical shifts, coupling constants, line widths) were specified as variable. The parameters are collected in Tables 2 and 3.
| [Hg3(μ-dppm)3](O3SCF3)4 | 4 | 1a | |||
|---|---|---|---|---|---|
| a Standard deviations for chemical shifts, P–P and Hg–P coupling constants calculated by the WIN-DAISY automatic routine are ≤0.1 Hz. b For the numbering scheme of Hg and P atoms see Scheme 1. c For designation of P–P and Hg–P couplings see Schemes 5 and 6. | |||||
| δ(P) | 54.6 | 54.8 | 97.6 | 90.8 | 61.2 (P1/2), 56.4 (P3/6), 62.0 (P4/5) |
| J(PP)cis/Hz | 134.7 | 135.1 | 177.4 | 179.5 | 127.1 (P1P6), 127.0 (P4P5) |
| J(PP)cis–cis/Hz | 86.7 | 86.7 | 97.9 | 98.3 | 100.0 (P1P2), 83.7 (P3P4) |
| J(PP)cis–trans/Hz | 83.7 | 83.7 | 80.3 | 80.5 | 81.9 (P1P3), 87.7 (P1P5), 76.9 (P3P5) |
| J(PP)trans–trans/Hz | 22.1 | 22.0 | 19.6 | 19.8 | 20.5 (P1P4), 25.6 (P3P6) |
| δ(Hg) | 2576 | 2576 | 2577 | 2577 | 2571 (Hg1), 2489 (Hg3) |
| 1 J(HgP)/Hz | 1793 | 1793 | 2158 | 2167 | 1664 (Hg1P1), 1798 (Hg1P6), 1927 (Hg3P4) |
| J(HgP)cis/Hz | 194 | 194 | 159 | 164 | 156 (Hg1P2), 264 (Hg1P5), 189 (Hg3P3) |
| J(HgP)trans/Hz | 1279 | 1279 | 1269 | 1266 | 1305 (Hg1P3), 1208 (Hg1P4), 1194 (Hg3P1) |
| J(HgHg)/Hz | 16404 ± 63 (Hg1Hg3) | ||||
| Solvent | CD2Cl2 | CD2Cl2–MeOH (2/1) | CD2Cl2 | CD2Cl2–MeOH (2/1) | CD2Cl2–MeOH (2/1) |
| 2a | 2b | 3 | |
|---|---|---|---|
| a Standard deviations for chemical shifts, P–P and Hg–P coupling constants calculated by the WIN-DAISY automatic routine are ≤0.1 Hz. b For the numbering scheme of Hg and P atoms see Scheme 1. c For designation of P–P and Hg–P couplings see Schemes 5 and 6. | |||
| δ(P) | 61.7 (P1/4), 57.9 (P2/3) | 57.6 (P1/4), 60.3 (P2/3) | 59.6 |
| J(PP)cis/Hz | 137.7 (P2P3) | 138.3 (P2P3) | — |
| J(PP)cis–cis/Hz | 92.0 (P1P2) | 84.9 (P1P2) | 92.4 (P1P2) |
| J(PP)cis–trans/Hz | 73.7 (P1P3) | 67.5 (P1P3) | — |
| J(PP)trans–trans/Hz | 31.4 (P1P4) | 36.0 (P1P4) | — |
| δ(Hg) | 2600 (Hg1/3), 2385 (Hg2) | 2628(Hg1/3), 2321(Hg2) | 2412(Hg1/2), 2663(Hg3) |
| 1 J(HgP)/Hz | 1876 (Hg1P1), 2189 (Hg2P2) | 1882 (Hg1P1), 2401 (Hg2P2) | 2268 (Hg1P1) |
| J(HgP)cis/Hz | 275 (Hg1P2), 163 (Hg2P1) | 358 (Hg1P2), 144 (Hg2P1) | 234 (Hg2P1) |
| J(HgP)trans/Hz | 1206 (Hg1P3), 1602 (Hg1P4) | 1149 (Hg1P3), 1684 (Hg1P4) | 1559 (Hg3P1) |
| J(HgHg)/Hz | 17361 ± 92 (Hg1Hg2) | ||
| 23499 ± 4 (Hg1Hg3) | |||
| Solvent | CD2Cl2–MeOH (2/1) | CD2Cl2–MeOH (2/1) | CD2Cl2–MeOH (2/1) |
The 31P{1H} spectrum of 1a may be divided into three subspectra: [ABM]2 (isotopomer without 199Hg nuclei), [ABM]2X (199Hg3) and AA′BB′MM′X (199Hg1 or 199Hg2). Signals arising from subspectra of the isotopomers containing more than one 199Hg are observed in the 31P{1H} NMR spectrum but corresponding intensities are too weak for iteration. Although there is a strong overlap of the patterns of the three subspectra the [ABM]2-part could be iterated independently from the other subspectra. By use of these data it was possible to iterate the whole spectrum including all P–P and Hg–P couplings. The assignment of the three sets of phosphorus atoms is based on comparison of the P–P and Hg–P coupling constants with the previously reported compounds [Hg3(μ-dpam)(μ-dppm)2](O3SCF3)4, [Hg3(μ-dpam)2(μ-dppm)](O3SCF3)4, [Hg3(μ-dpam)2(μ-Ph2AsCH2PPh2)](O3SCF3)4 and [Hg3(μ-Ph2AsCH2PPh2)3](O3SCF3)4.7d
The diastereomeric compounds 2a and 2b show two [AB]2-systems (isotopomers without 199Hg, ratio 2/1) in the 31P{1H} NMR spectrum. The assignment of the chemical shifts of P1/P4 and P2/P3 is clearly proved by the [AB]2X and AA′BB′X subspectra in the 31P{1H} NMR spectrum (isotopomers containing one 199Hg). The assignment of the 31P and 199Hg NMR parameters are consistent with the appearance of an A2 and an AB system in the ratio 2/1 for the dpam methylene protons in the 1H NMR spectrum of 2a and 2b.
The 31P{1H} spectrum of 4 shows a singlet attributable to the [[A]2]3 spin system of the isotopomer without 199Hg nuclei flanked by widespread mercury satellites (spin system [AA′A″]2X). The satellite patterns attributable to the three different Hg–P coupling constants are well separated. Only regions of the satellite spectrum which are not superimposed by the central singlet were used for the iteration. The spectrum shows also lines of weak intensity, arising from the isotopomer containing two active 199Hg, which were not considered for iteration.
The spin systems of the 31P{1H} spectrum of [Hg3(μ-dppm)3](O3SCF3)4 are the same as for 4, but the analysis is more difficult because similar values of 1J(HgP) and J(HgP)trans cause an overlap of the corresponding patterns of the isotopomer containing one 199Hg nucleus. The analysis of [Hg3(μ-dppm)3](O3SCF3)4 succeeded by use of the parameters of 1a and 4 as starting values. The P–P coupling constants exhibiting the highest and the smallest value were assigned to J(PP)cis and J(PP)trans–trans, respectively.7d The assignment of J(PP)cis–cis and J(PP)cis–trans results conclusively from symmetry considerations, which were taken into account during the simulation: J(PP)cis–trans is the only coupling, which appears six times in the complex cation, whereas all other couplings appear only three times. The 31P and 199Hg NMR parameters of [Hg3(μ-dppm)3](O3SCF3)4 presented here are the result of the first complete and successful NMR spectroscopic analysis of a [M3(μ-dppm)3] system.
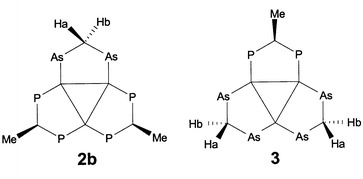
The 199Hg{1H} spectra (see Fig. 3 for the 199Hg{1H} spectrum of [Hg3(μ-dppm)3](O3SCF3)4) of all compounds were simulated by use of the parameters derived from their 31P{1H} spectra. For compound 1a a 199Hg{31P,1H} NMR spectrum has been recorded in order to determine the Hg1–Hg3 coupling constant from the AB subspectrum of the isotopomer containing two active 199Hg nuclei. The Hg–Hg coupling constants of 3 have been extracted from the [AX]2 and AA′XY 199Hg subspectra of the isotopomers containing two active 199Hg nuclei, respectively.
4 (center at 2576 ppm, spectral width 7300 Hz). Small differences in intensity and lineshape are due to subspectra with more than one active 199Hg in the experimental spectrum, which were not considered in the simulation.](/image/article/2002/DT/b102840p/b102840p-f3.gif) | ||
| Fig. 3 Experimental (upper trace) and simulated (lower trace) 199Hg{1H} NMR spectrum of [Hg3(μ-dppm)3](O3SCF3)4 (center at 2576 ppm, spectral width 7300 Hz). Small differences in intensity and lineshape are due to subspectra with more than one active 199Hg in the experimental spectrum, which were not considered in the simulation. | ||
Discussion of specific 31P and 199Hg NMR parameters
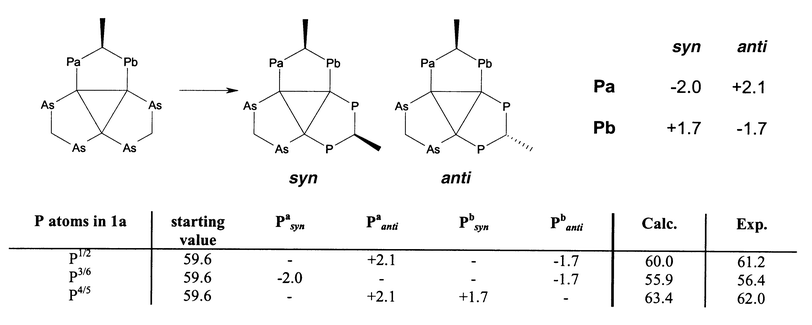 | ||
| Scheme 7 Effect of the substitution of one dpam ligand in compound 3 by mdppm on the 31P shift and calculation of the 31P shifts of 1a by the increments obtained (all values in ppm). | ||
The 31P shift of 4 is remarkably solvent dependent and shows values of 97.6 ppm (CH2Cl2) or 90.8 ppm (CH2Cl2–MeOH, 2/1), respectively.
The 199Hg shifts of all new compounds reported in this article are found in the characteristic range of [Hg3]4+ clusters, exhibiting values markedly downfield relative to arsine or phosphine complexes of mercury(II).7d The recently described substituent perturbations for δ(Hg) when replacing an arsine by a phosphine ligand (large positive influence of the trans position, smaller negative influence of the ipso and cis positions, respectively)7d are found in the mdppm derivatives of [Hg3]4+ as well as in the related dppm and Ph2PCH2AsPh2 complexes, but there exists an additional dependence on the relative orientations of the methyl groups (syn, anti): Mercury atoms co-ordinated by two mdppm phosphorus atoms of the ligands in syn orientation feature a 199Hg shift distinctly upfield compared to their anti analogues. The Hg resonances of the compounds [Hg3(μ-dppm)3](O3SCF3)4 and 4 exhibit nearly the same values (in contrast to Hg2 in 1a). This may indicate that δ(Hg) is predominantly influenced by steric and not by electronic factors for [Hg3]4+ complexes surrounded by the same atomic donor set.
Comparing 1J(HgP) in the compounds [Hg3(μ-dppm)3](O3SCF3)4 (1793 Hz), 1a (1796 Hz; mean value) and 4 (2167 Hz), which all bear six phosphorus ligands, the value of 4 containing dppa is markedly increased. It is well known that phosphite complexes of mercury(II) exhibit higher 1J(HgP) values than their phosphine analogues. The substitution of a phosphorus bound carbon atom by a more electronegative element, e.g. N, O, F or Cl, generally leads to larger 1J(HgP) coupling constants.20 As one can easily recognise, merely the one-bond Hg–P coupling constant is affected by several hundreds of Hz due to the complete substitution of dppm in [Hg3(μ-dppm)3](O3SCF3)4 by dppa in 4. Since all other couplings are only slightly changed, this corresponds to a quasi selective variation of one Hg–P coupling constant.
The different Hg–P couplings in [Hg3]4+ complexes involving the ligands dpam, dppa, dppm, mdppm and Ph2PCH2AsPh2 generally decrease in the order 1J(HgP) > J(HgP)trans ≫ J(HgP)cis, although there is a slight overlap between 1J(HgP) and J(HgP)trans (Fig. 4).
![Ranges for Hg–P couplings in [Hg3]4+ complexes.](/image/article/2002/DT/b102840p/b102840p-f4.gif) | ||
| Fig. 4 Ranges for Hg–P couplings in [Hg3]4+ complexes. | ||
The value of J(PP)cis in compound 4 (177.4 Hz) is slightly offset against the values found for 1a, 2a, 2b, [Hg3(μ-dppm)3](O3SCF3)4 and [Hg3(μ-dpam)(μ-dppm)2](O3SCF3)4 (146.9–127.0 Hz). This may be caused by the same factors discussed for 1J(HgP) couplings.20
In the complexes [Hg3(μ-dppm)3](O3SCF3)4, 1a, 2a, 2b and 4 the values of J(PP)cis–trans range between 87.7 and 67.5 Hz. If mdppm ligands are in a syn orientation the coupling constant seems to be higher than for mdppm ligands in an anti orientation. The substitution of a dppm or mdppm ligand in [Hg3(μ-dppm)3](O3SCF3)4 or 1a by dpam, leads to an increase of J(PP)trans–trans.
The value of J(PP)trans–trans in compound 2a (anti) is higher than in 2b (syn). This effect is also observed for the mdppm ligands in syn and anti arrangements in compound 1a.
The values for J(PP)cis and J(PP)trans–trans in [Hg3]4+ complexes involving the ligands dpam, dppa, dppm, mdppm and Ph2PCH2AsPh2 exhibit distinct ranges, which are well separated from other P–P couplings, J(PP)cis–cis and J(PP)cis–trans exhibit similar values, but generally cis–trans are smaller than cis–cis coupling constants (Fig. 5).
![Ranges for P–P couplings in [Hg3]4+ complexes.](/image/article/2002/DT/b102840p/b102840p-f5.gif) | ||
| Fig. 5 Ranges for P–P couplings in [Hg3]4+ complexes. | ||
The values of J(PP)cis–trans and J(PP)trans–trans are intriguing because trans–trans are smaller than cis–trans coupling constants. This observation clearly emphasises that qualitative arguments like cis and trans relations, which are well established tools for the interpretation of magnitudes of coupling constants in transition metal co-ordination compounds, may not be valid for cyclic structures with unusual bonding geometries. Undoubtedly, there is still some theoretical investigation needed to rationalise the coupling constants in [Hg3]4+ clusters.
Supposing that 1J(HgP) coupling constants have a positive sign,21 all Hg–P and P–P coupling constants derived from the 31P{1H} second order subspectra of the compounds [Hg3(μ-dppm)3](O3SCF3)4, 1a, 2a, 2b and 4 are positive. In view of this result, we propose positive values for all Hg–P and P–P coupling constants of the various [Hg3]4+ clusters reported in this article.
Single crystal X-ray structure of 4
Compound 4 crystallises from CHCl3 as colourless prisms containing four solvent molecules per formula unit. An ORTEP32 plot of the structure is shown in Fig. 6. The [Hg3(μ-dppa)3]4+ cation acts as a bifunctional recognition host as has been reported for the [Hg3(μ-dppm)3]4+ system7a,c and two of the O3SCF3− anions are located inside the two cavities formed by the 12 phenyl groups of the dppa ligands and the Hg3 triangle. One anion is connected via a hydrogen bond to the nitrogen atom of one of the three dppa ligands. The fourth anion bridges between two [Hg3(μ-dppa)3]4+ cations via two O⋯H⋯N hydrogen bonds (O10 and O12) resulting in an infinite chain structure.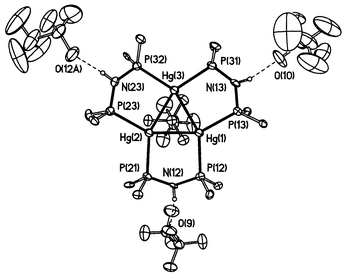 | ||
| Fig. 6 Molecular structure of 4. One trifluoromethanesulfonate anion placed above the Hg3 plane has been omitted for clarity, only the ipso carbon atoms of the dppa-bound phenyl groups are shown. The trifluoromethanesulfonate anion containing O(12A) was generated by a symmetry operation from the trifluoromethanesulfonate anion containing O(10). | ||
The geometry of the [Hg3(μ-dppa)3]4+ system is similar to that of [Hg3(μ-dppm)3]4+: Three dppa ligands bridge the edges of the Hg3 triangle. The Hg–Hg distances are 278.71–282.35 pm compared with 271.7–286.49 pm observed for various [Hg3(μ-dppm)3]4+ clusters.7a,c The Hg–P distances are 250.6–253.6 pm. The P–N distances (165.3–169.2 pm) show values between a P–N single bond (177 pm) and a P–N double bond (156 pm) in phosphacenes as has been reported for other dppa complexes.22 The five-membered Hg2P2N rings formed by the bridging dppa ligands and one edge of the Hg3 triangle adopt envelope conformations with the nitrogen atoms at the flap. Just as observed for [Hg3(μ-dppm)3]4+ clusters, two flaps are oriented below the Hg3 plane and one above. Two of the Hg–O separations are within the lower Hg–O van der Waals limit of 290 pm based on a Hg radius of 150 pm, and all Hg–O contacts are below the value of 313 pm based on a mercury radius of 173 pm. The N–H⋯O distances are 278.0–283.9 pm. Hydrogen bond interactions concerning anions or solvent molecules, respectively, were previously observed in various dppa complexes.23 In compound 4 two of the hydrogen atoms attached to the nitrogens are found to lie distinctly out of the PNP plane (H12N: 33(9) pm; H23N: 32(12) pm), and this is thought to be caused by the interaction with the anions.
Conclusion and outlook
We succeeded in demonstrating that an unambiguous characterisation of [Hg3(μ-dppm)3](O3SCF3)4 and related subvalent [Hg3]4+ clusters in solution is possible by multinuclear NMR spectroscopy. In comparison with the single crystal X-ray data in the solid state, we obtained additional information about the conformation dynamics in these systems. The observed parameters display, in part, unexpected values, which still need theoretical investigations to be rationalised.The different thermodynamic stabilities of diastereomeric mdppm bridged [Hg3]4+ complexes could be attributed to the rigid C-envelope conformation of the five-membered rings formed by two metal atoms and the P–C–P backbone of the ligand with the methyl group oriented in the less sterically crowded equatorial position. This explanation may also be valid for other complexes containing more than one bridging mdppm ligand.12–14
Complete exchange of dppm by mdppm in [M3(μ-dppm)3] type compounds seems to be a promising derivatisation technique for such systems in order to obtain information about their 31P NMR parameters. The preferential formation of the less symmetrical syn–anti–anti isomer and the comparatively large effects of the relative ligand orientation on the 31P shift are a good basis for getting spectra, which are accessible for partial analysis and which may in turn be successfully simulated.
The synthesis of mixed ligand diarsine–diphosphine complexes might also be a good tool for making accessible 31P NMR parameters for [M3(μ-dppm)3] compounds or higher nuclearity clusters bearing chemically equivalent dppm ligands. Especially for [Hg3]4+ the existence of mixed ligand compounds could a priori not be predicted, because it is well known that mercury complexes bearing different ligands often undergo symmetrisation reactions.
The more pronounced influence of substituents at the phosphorus atom on 1J(HgP) coupling constants compared to couplings involving more than one bond might be a more general phenomenon for M–P couplings in transition metal clusters. There are certainly extensive investigations necessary to verify this thesis.
The interpretation of our 1H NMR spectroscopic results concerning the protons at the envelope carbon atom of the ligand backbone can help to rationalise 1H NMR data found for other [M3(μ-dppm)3] type compounds.
At least we propose [Hg3]4+ clusters being interesting subjects for 199Hg solid state NMR spectroscopy, which has received increasing interest in the past few years: Extensive investigations have been performed for Hg(II) compounds,24 two reports involve Hg(I).25 Solid state NMR spectroscopy should yield additional information to solution NMR data which could be used to probe structure and bonding. For example, determination of the 199Hg shielding tensor for a variety of [Hg3]4+ compounds could give insights into orbitals contributing to the bonding in such compounds. Besides the subvalent mercury clusters stabilised by bridging phosphine and arsine ligands, there are two examples of this class of compounds existing merely in a crystal lattice, these are the mineral terlinguait, Hg4Cl2O2, and Hg9As4O16.26,27 For these two species solid state NMR spectroscopy represents the sole method for getting information about their 199Hg data.
Experimental
Materials
[Hg(Me2SO)6](O3SCF3)2, dpam and mdppm were prepared according to published procedures.13b,28,29 The syntheses of [Hg3(μ-dppm)3](O3SCF3)4 and [Hg3(μ-dpam)3](O3SCF3)4 have been described previously.7a,d All other chemicals were purchased from commercial suppliers and used without further purification. Elemental analyses were performed by the Institut für Physikalische Chemie, Universität Wien.Syntheses
NMR spectroscopy
199Hg{1H}, 31P{1H} and 1H NMR spectra were recorded at 20 °C on Bruker DPX 300 or DRX 500 spectrometers. All spectra were recorded using Bruker standard pulse programs, the 199Hg{31P,1H} spectrum of compound 1a was recorded on a Bruker DRX 500 spectrometer by WALTZ 16 decoupling in the spectral region of the 31P signals. The connectivities of the methylene protons in compounds 2b and 3 could be established unambiguously by 1H–1H-COSY spectra. Free induction decays were processed by exponential multiplication (LB-values: 20 [199Hg], 2 [31P] or 0.1 [1H], respectively) before Fourier transformation, the baselines of the spectra were smoothed by manual baseline correction prior to iteration. Errors in chemical shifts and coupling constants were calculated by the WIN-DAISY automatic routine; these are statistical in nature and probably underestimate the true errors, which also depend on systematic and experimental contributions. 31P/199Hg chemical shifts are reported relative to 85% H3PO4/2 mmol HgO in 1 ml 60% HClO4, used as an external standard, 1H chemical shifts are relative to Me4Si and were determined by reference to the residual 1H solvent peaks. Coupling constants are reported in Hz.Crystallography
Crystals of [Hg3(μ-dppa)3](O3SCF3)4·4CHCl3 were obtained upon slow evaporation of a solution of [Hg3(μ-dppa)3](O3SCF3)4 in CHCl3 at ambient temperature. A colourless prismatic crystal with dimensions 0.7 × 0.65 × 0.5 mm was mounted on a glass fibre, X-ray data were collected at 213(2) K on a Siemens P4 diffractometer (Mo-Kα radiation, monochromator: Highly oriented graphite crystal, ω-scan method). Unit cell parameters were determined and refined from 30 randomly selected reflections in the θ-range between 5.3 and 12.5°, obtained by P4 automatic routine. Crystal system: Monoclinic, space group: P21/n (no. 14), unit cell dimensions: a = 1880.9(4), b = 2190.0(9), c = 2508.0(5) pm, α = 90°, β = 95.81(2)°, γ = 90°. 9668 reflections—8684 of which were independent (Rint = 0.0347)—were collected in the θ-range between 3.04 and 19.99° (index ranges: 0 ≤ h ≤ 18, −1 ≤ k ≤ 21, −22 ≤ l ≤ 22). Every 97 reflections 3 standard reflections were measured. Data were corrected for Lorentz-polarisation and absorption effects (ψ-scans). The structure was solved by direct methods and subsequent difference Fourier techniques (SHELXS-86),30 refinement was carried out by full-matrix least-squares methods (SHELXL-93).31 All non-hydrogen atoms were refined anisotropically. The amine hydrogen atoms were fixed to a distance of 85 pm and refined with isotropic parameters, all other hydrogen atoms were placed at calculated ideal positions (riding model). One solvent molecule is disordered about the C8–H8 axis over two positions (Cl10–Cl12/Cl1A–Cl3A) with occupancies 2/1. Final R indices are R1 = 0.0360, wR2 = 0.0752 (I > 2σ(I)) and R1 = 0.0581, wR2 = 0.0865 (all data).CCDC reference number 154492.
See http://www.rsc.org/suppdata/dt/b1/b102840p/ for crystallographic data in CIF or other electronic format.
Acknowledgements
Financial support from the Fonds zur Förderung der wissenschaftlichen Forschung, Project P 11842-PHY is gratefully acknowledged.References
- (a) D. A. Morgenstern, R. E. Wittrig, P. E. Fanwick and C. P. Kubiak, J. Am. Chem. Soc., 1993, 115, 6470 CrossRef CAS; (b) D. A. Morgenstern, C. C. Bonham, A. P. Rothwell, K. V. Wood and C. P. Kubiak, Polyhedron, 1995, 14, 1129 CrossRef CAS.
- (a) L. Manojlovic-Muir, K. W. Muir, B. R. Lloyd and R. J. Puddephatt, J. Chem. Soc., Chem. Commun., 1983, 1336 RSC; (b) R. Provencher, K. T. Aye, M. Drouin, J. Gagnon, N. Boudreault and P. D. Harvey, Inorg. Chem., 1994, 33, 3689 CrossRef CAS; (c) T. Zhang, M. Drouin and P. D. Harvey, Chem. Commun., 1996, 877 RSC; (d) P. D. Harvey, K. Hierso, P. Braunstein and X. Morise, Inorg. Chim. Acta, 1996, 250, 337 CrossRef CAS.
- (a) G. Ferguson, B. R. Lloyd and R. J. Puddephatt, Organometallics, 1986, 5, 344 CrossRef CAS; (b) R. J. Puddephatt, L. Manojlovic-Muir and K. W. Muir, Polyhedron, 1990, 9, 2767 CrossRef CAS; (c) B. T. Sterenberg, M. C. Jennings and R. J. Puddephatt, Organometallics, 1999, 18, 3737 CrossRef CAS.
- (a) J. K. Bera, M. Nethanji and A. G. Samuelson, Inorg. Chem., 1999, 38, 218 CrossRef CAS; (b) J. K. Bera, M. Nethanji and A. G. Samuelson, Inorg. Chem., 1999, 38, 1725 CrossRef CAS; (c) B. F. Straub, F. Rominger and P. Hofmann, Inorg. Chem., 2000, 39, 2113 CrossRef CAS.
- (a) A. A. M. Aly, D. Neugebauer, O. Orama, U. Schubert and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1978, 17, 125 CrossRef; (b) D. Franzoni, G. Pelizzi, G. Predieri, P. Tarasconi, F. Vitali and C. Pelizzi, J. Chem. Soc., Dalton Trans., 1989, 247 RSC; (c) V. W.-W. Yam, W. K.-M. Fung and K.-K. Cheung, Chem. Commun., 1997, 963 RSC; (d) V. G. Albano, C. Castellari, M. C. Iapalucci, G. Longoni, M. Monari, A. Paselli and S. Zacchini, J. Organomet. Chem., 1999, 573, 261 CrossRef CAS.
- J. W. A. van der Velden, J. J. Bour, R. Pet, W. P. Bosman and J. H. Noordik, Inorg. Chem., 1983, 22, 3112 CrossRef CAS.
- (a) B. Hämmerle, E. P. Müller, D. L. Wilkinson, G. Müller and P. Peringer, J. Chem. Soc., Chem. Commun., 1989, 1527 RSC; (b) A. Knoepfler, E. Ellmerer-Müller, K.-H. Ongania, K. Wurst and P. Peringer, J. Chem. Soc., Dalton Trans., 1997, 1607 RSC; (c) A. Knoepfler-Mühlecker, B. Scheffter, H. Kopacka, K. Wurst and P. Peringer, J. Chem. Soc., Dalton Trans., 1999, 2525 RSC; (d) A. Knoepfler-Mühlecker, W. Schuh, B. Scheffter, H. Kopacka, K. Wurst and P. Peringer, Inorg. Chim. Acta, 2000, 303, 70 CrossRef CAS; (e) B. Scheffter, W. Schuh, K.-H. Ongania, H. Kopacka, R. Malleier, K. Wurst and P. Peringer, Polyhedron, 2000, 19, 871 CrossRef CAS.
- J. T. Mague, J. Cluster Sci., 1995, 6, 217 CAS.
- (a) W. R. Mason, Inorg. Chem., 1997, 36, 1167; (b) H. Kunkely and A. Vogler, Chem. Phys. Lett., 1993, 206, 467 CrossRef CAS.
- P. Braunstein, R. Devenish, P. Gallezot, B. T. Heaton, C. J. Humphreys, J. Kervennal, S. Mulley and M. Ries, Angew. Chem., Int. Ed. Engl., 1988, 27, 927 CrossRef.
- P. Braunstein, C. de Méric de Bellefon and M. Ries, Inorg. Chem., 1988, 27, 1338 CrossRef CAS.
- P. A. W. Dean and R. S. Srivastava, Can. J. Chem., 1985, 63, 2829 CAS.
- (a) G. Besenyei, C.-L. Lee, Y. Xie and B. R. James, Inorg. Chem., 1991, 30, 2446 CrossRef CAS; (b) C.-L. Lee, Y.-P. Yang, S. J. Rettig, B. R. James, D. A. Nelson and M. A. Lilga, Organometallics, 1986, 5, 2220 CrossRef CAS.
- A. F. M. J. van der Ploeg and G. van Koten, Inorg. Chim. Acta, 1981, 51, 225 CrossRef CAS.
- (a) F. Neve, M. Ghedini, A. Tiripicchio and F. Ugozzoli, Organometallics, 1992, 11, 795 CrossRef CAS; (b) G. J. Arsenault, L. Manojlovic-Muir, K. W. Muir, R. J. Puddephatt and I. Teurnicht, Angew. Chem., Int. Ed. Engl., 1987, 26, 86 CrossRef; (c) A. T. Hutton, C. R. Langrick, D. M. McEwan, P. G. Pringle and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1985, 2121 RSC; (d) R. J. Puddephatt and M. A. Thomson, Inorg. Chem., 1982, 21, 725 CrossRef CAS; (e) A. Blagg, A. T. Hutton, P. G. Pringle and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1984, 1815 RSC.
- (a) F. R. Jensen, D. S. Noyce, C. H. Sederholm and A. J. Berlin, J. Am. Chem. Soc., 1960, 82, 1256 CrossRef CAS; (b) A. G. Moritz and N. Sheppard, Mol. Phys., 1962, 5, 361 CAS.
- (a) U. Weber, R. Spiske, H.-W. Höffken, G. Hägele and H. Thiele, Manual and Program system, Bruker Manual, 1993 Search PubMed; (b) G. Hägele, P. Reinemer and M. Grzonka, Workshop “Computer in der Chemie”, Software-Entwicklung in der Chemie, 1988, vol. 2, p. 241 Search PubMed.
- (a) R. J. Gillespie, P. Granger, K. R. Morgan and G. J. Schrobilgen, Inorg. Chem., 1984, 23, 887 CrossRef CAS; (b) R. Malleier, H. Kopacka, W. Schuh, K. Wurst and P. Peringer, Chem. Commun., 2001, 51 RSC.
- (a) G. B. Deacon, M. J. O'Connor and G. N. Stretton, Aust. J. Chem., 1986, 39, 953 CAS; (b) J. Eichbichler and P. Peringer, Chem. Ber., 1984, 117, 1215 Search PubMed; (c) P. A. W. Dean, J. J. Vittal and M. H. Trattner, Inorg. Chem., 1987, 26, 4245 CrossRef CAS.
- C. J. Jameson, in Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis, eds. J. G. Verkade and L. D. Quin, VCH, Deerfield Beach, FL, 1987, p. 205 Search PubMed.
- (a) I. J. Colquhoun and W. McFarlane, J. Chem. Soc., Dalton Trans., 1981, 658 RSC; (b) A. Handler, P. Peringer and E. P. Müller, J. Organomet. Chem., 1990, 389, C23 CrossRef CAS; (c) C. A. Ghilardi, S. Midollini, S. Moneti, A. Orlandini and J. A. Ramirez, J. Chem. Soc., Chem. Commun., 1989, 304 RSC.
- N. N. Greenwood and A. Earnshaw, Chemie der Elemente, VCH, Weinheim, 1988, p. 698 Search PubMed.
- (a) F. A. Knoch and K. J. Meier, Z. Naturforsch., Teil B, 1991, 46, 1699 Search PubMed; (b) J. Ellermann, F. A. Knoch and K. J. Meier, Z. Naturforsch., Teil B, 1990, 45, 1657 Search PubMed; (c) J. Ellermann, F. A. Knoch, K. J. Meier and M. Moll, J. Organomet. Chem., 1992, 428, C44 CrossRef CAS; (d) M. Knorr and C. Strohmann, Eur. J. Inorg. Chem., 1998, 495 CrossRef CAS.
- G. A. Bowmaker, R. K. Harris and S.-W. Oh, Coord. Chem. Rev., 1997, 167, 49 CrossRef CAS.
- (a) G. A. Bowmaker, R. K. Harris and D. C. Apperley, Inorg. Chem., 1999, 38, 4956 CrossRef CAS; (b) R. A. Santos and G. S. Harbison, J. Am. Chem. Soc., 1994, 116, 3075 CrossRef CAS.
- (a) S. Scavnicar, Acta Crystallogr., 1956, 9, 956 CrossRef; (b) K. Aurivillius and L. Folkmarson, Acta Chem. Scand., 1968, 22, 2529 Search PubMed; (c) K. Brodersen, G. Göbel and G. Liehr, Z. Anorg. Allg. Chem., 1989, 575, 145 CrossRef CAS.
- A. L. Wessels, W. Jeitschko and M. H. Möller, Z. Naturforsch., Teil B, 1997, 52, 469 Search PubMed.
- P. Peringer, J. Inorg. Nucl. Chem., 1980, 42, 1501 Search PubMed.
- A. M. Aguiar, J. T. Mague, H. J. Aguiar, T. G. Archibald and B. Preiean, J. Org. Chem., 1968, 33, 1681 CrossRef CAS.
- G. M. Sheldrick, SHELXS-86: Program for crystal structure solutions, Göttingen, 1986 Search PubMed.
- G. M. Sheldrick, SHELXL-93: Program for refinement of crystal structures, Göttingen, 1993 Search PubMed.
- C. K. Johnson, ORTEP, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, 1976 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |