Synthesis, structure and reactivity of 1-(α-C,α′-halo-o-xylyl)-2-trialkylsilyl-1,2-dicarbaboranes†
Andrei S.
Batsanov
,
Charlotte K.
Broder
,
Andrés E.
Goeta
,
Judith A. K.
Howard
,
Andrew K.
Hughes
* and
John M.
Malget
Department of Chemistry, University Science Laboratories, South Road, Durham, UK DH1 3LE. E-mail: a.k.hughes@durham.ac.uk
First published on 3rd December 2001
Abstract
Treating Li[tBuMe2Si-1,2-C2B10H10] with excess α,α′-dihalo-o-xylenes, 1,2-C6H4(CH2X)2 (X = Br, Cl), generates only 1-(α-C,α′-halo-o-xylyl)-2-(tert-butyldimethylsilyl)-1,2-dicarba-closo-dodecaboranes, 1-{o-(XCH2C6H4CH2)}-2-tBuMe2Si-1,2-C2B10H10 (X = Cl 1a, Br 1b). The structures of both 1a and 1b were determined by single crystal X-ray diffraction. Reaction of either 1a or 1b with nBuLi or MeLi affords the substituted ethane, (2-tBuMe2Si-1,2-C2B10H10-1-o-CH2C6H4)2C2H42 whereas reaction with tBuHNLi affords the substituted ethene (2-tBuMe2Si-1,2-C2B10H10-1-o-CH2C6H4)2C2H23; both structures were confirmed by X-ray diffraction. Cleavage of the carborane–silicon bond in 1a or 1b by Bu4NF gives dihydronaphthocarborane, 4, which has been structurally characterised.
Introduction
Functionalised cyclopentadienyl ligands have made a significant impact on the chemistry of the early transition metals in recent years through the high activity of constrained geometry cyclopentadienyl amide ligands in alkene polymerisation.1 Pendant arm donor ligand cyclopentadienyls have also been of significant interest in the chemistry of main group and other transition metals.2,3 Given the relationship between cyclopentadienyl ligands and the nido-C2B9H11 ligand,4 we5 and others6,7 have been exploring the synthesis of ligands containing nido-carboranes attached to cyclopentadienyl,8 amine and other donor atoms through carbon chains of various types and lengths. The development of bio-compatible amine-functionalised carboranes for a variety of other purposes, including use in boron neutron capture therapy (BNCT) remains an active area of research.9We5 have recently reported the synthesis of closo- and nido-carboranylamines via modifications to known procedures, and were interested to explore other syntheses. Thus the displacement of a tosyl or halide group by nucleophilic azide ion10 and subsequent reduction to the amine11 is a well-established procedure.12 It has previously been reported however that this reaction fails for (halomethyl)carboranes, XCH2C2B10H11, X = Cl, Br, and only proceeds for the iodo derivatives.13 Similar failures have been reported for a variety of reactions by other groups and were rationalised by the electronic influence of the carborane.14
The reactions of carborane nucleophiles with α,α′-dihalo-o-xylenes are well-established. The di-lithiation of o-carborane gives Li2C2B10H10, which reacts with α,α′-dibromo-o-xylene to give dihydronaphthocarborane, a precursor to naphthocarborane.15 Mono-lithiation of o-carborane is not always a clean reaction, but the same effect is achieved by mono-silylation,16–18 prior to metallation and Hawthorne reports that the reaction of two equivalents of Li(tBuMe2SiC2B10H10) with α,α′-dihalo-o-xylenes gives substitution of both halides by the large carborane nucleophile (Scheme 1).17
 | ||
| Scheme 1 Product of the 2 ∶ 1 reaction reported by Hawthorne. | ||
We decided to explore the reaction of one equivalent of Li(tBuMe2SiC2B10H10) with α,α′-dihalo-o-xylenes as a route to the synthesis of carborane–xylyl–amine ligands since we reasoned that carborane–xylyl–halide intermediates would be amenable to nucleophilic displacement. Here we report that these compounds also exhibit inactivity to nucleophilic substitution that in retrospect may be exploited to synthetic advantage to allow access to novel molecular architectures.
Results and discussion
The reaction of one equivalent of Li(tBuMe2SiC2B10H10) with an excess of α,α′-dihalo-o-xylenes results in mono-substitution and affords 1-(α-C,α′-halo-o-xylyl)-2-(tert-butyldimethylsilyl)-1,2-dicarba-closo-dodecaboranes, 2-tBuMe2Si-1-{o-(XCH2C6H4CH2)}-1,2-C2B10H10 (X = Cl, 1a; Br; 1b) in excellent yield (Scheme 2). In keeping with the α,α′-dihalo-o-xylenes from which they are derived these compounds are potent irritants and lachrymators and as such skin contact should be avoided. Spectroscopic data for the Cl (1a) and Br (1b) derivatives are similar, so that only the chloro-derivative will be discussed. Considering the 1H NMR data, the tBuMe2Si substituent is clearly observed δ 0.46 (s, 6H, SiMe), 1.15 (s, 9H, Bu), and both methylene linkages are chemically distinct δ 3.70 (s, 2H, CH2), 4.66 (s, 2H, CH2). Due to the chemical inequivalence of the two methylene moieties the C6H4 unit is a complex multiplet between δ 7.18 and 7.39. The 11B NMR spectrum confirms the retention of the closo-C2B10 framework. Retention of the silyl protective group lends useful solubility and crystallinity which greatly aids separation and purification. Single crystal X-ray diffraction analysis for both 1a and 1b serves to further confirm the structural features with all parameters well within the expected range for compounds of this type. Fig. 1 shows the molecular structure of 1a, that of 1b is essentially identical. Selected bond lengths and angles appear in Table 1 and reveal C(1)–C(2) distances of 1.699(2) Å for 1a and 1.697(4) Å for 1b, which, although long for C–C single bonds, are within the normal range for polyhedral closo-carboranes, where the carbon atoms have large coordination numbers, here six. The other metric parameters are unremarkable.| 1a | 1b | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| X = Cl(1) | X = Br(1) | X = C(18A) | X = C(18A) | X = C(2) | |
| C(1)–C(2) | 1.699(2) | 1.697(4) | 1.694(2) | 1.702(2) | 1.649(2) |
| C(2)–Si(1) | 1.9652(14) | 1.961(3) | 1.960(2) | 1.9581(13) | |
| C(1)–C(11) | 1.547(2) | 1.540(4) | 1.549(2) | 1.542(2) | 1.519(2) |
| C(11)–C(12) | 1.521(2) | 1.526(4) | 1.517(2) | 1.518(2) | 1.518(2) |
| C(18)–X | 1.825(2) | 1.981(3) | 1.549(3) | 1.334(2) | 1.528(2) |
| C(1)–C(2)–Si(1) | 123.36(9) | 123.6(2) | 124.91(9) | 124.01(8) | |
| C(18)–C(2)–C(1) | 117.41(11) | ||||
| C(2)–C(1)–C(11) | 117.06(11) | 116.8(2) | 116.51(12) | 117.63(9) | 117.71(11) |
| C(1)–C(11)–C(12) | 114.49(11) | 114.8(2) | 113.75(12) | 114.47(10) | 114.97(12) |
| C(17)–C(18)–X | 110.14(10) | 109.4(2) | 111.3(2) | 125.7(2) | 114.96(12) |
 | ||
| Fig. 1 A view of the molecular structure of 1a showing 50% probability ellipsoids, hydrogen atoms as arbitrary sized spheres. | ||
 | ||
| Scheme 2 Reactions discussed in this work. Reagents and conditions: (i) nBuLi, THF, room temp; (ii) tBuHNLi, THF, −100 °C; (iii) nBu4NF, THF, −78 °C. | ||
In keeping with previous studies, we find these compounds are either inert to or are decomposed by reagents typically used to effect conversion of a halo group to an amine. Reagents investigated included NaN3 and hexamethylenetetraamine (Delépine reaction).19 No product of the desired formulation was obtained, as might be expected under such harsh conditions given that o-carborane is readily decapitated by amines.20 Thus, whilst compounds 1a and 1b are potential precursors to functionalised closo- and nido-carboranes, they are not suitable for the synthesis of such carboranes carrying basic, or other, functions which are capable of deboronating 1a and 1b in competition with nucleophilic substitution of the chloride or bromide.
Given these limitations it is of interest to establish what transformations are possible for such systems. Carbarods21 derived from carboranes are attracting attention as potential one-dimensional conductors and some of these materials exhibit liquid crystal behaviour.22 For these reasons it was of interest to establish whether the halomethylene unit would undergo coupling reactions, as such a methodology could be applied to the rational stepwise construction of oligomeric materials.23 Reaction of either 1a or 1b with nBuLi or MeLi results in a Wurtz type coupling reaction to afford the substituted ethane (2-tBuMe2Si-1,2-C2B10H10-1-o-CH2C6H4)2C2H4 (2) in reasonable yield as a colourless highly crystalline air-stable solid. The formulation is entirely consistent with spectroscopic and analytical data and is firmly established by a single crystal X-ray diffraction study. Considering 1H NMR data, a large change in chemical shift is observed for the methylene unit following loss of the electronegative halogen substituent and coupling to form the ethane (δ 4.66 for CH2Cl in 1a, δ 2.96 for CH2CH2 in 2). In both 1H and 13C NMR data other characteristic resonances of both the aryl and BuMe2Si units remain largely unaffected by the transformation. The 11B NMR spectrum confirms retention of the closo-carborane fragment. The molecular structure determined by the single crystal X-ray study is shown in Fig. 2 and the structural parameters are reported in Table 1. The molecule sits on a crystallographic inversion centre at the mid-point of the C(18)–C(18A) bond, the length of which, 1.549(3) Å, is entirely consistent with a C–C single bond. The hybridisation of C(18) is sp3 as evidenced by the C(17)–C(18)–C(18A) angle of 111.3(2)°. Other distances fall within normal ranges.
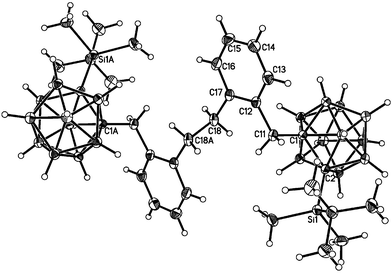 | ||
| Fig. 2 Molecular structure of alkane 2 showing 50% probability ellipsoids, hydrogen atoms as arbitrary sized spheres. Atoms labelled with the suffix “A” are generated by the symmetry operation (1 − x, −y, −z). | ||
In the light of the alkyl lithium-promoted coupling reaction, we sought to discover if alternative bridge functionality could be generated. Treatment of either 1a or 1b with tBuHNLi results in bis-dehydrohalogenation to afford the alkene dimer trans-[2-tBuMe2Si-1,2-C2B10H10-1-o-CH2C6H4]2C2H23 in high yield as a colourless highly crystalline solid. Such a reaction is of particular synthetic utility as it provides a potential route to conjugated materials. Considering 1H NMR data for 3, the most notable feature is the resonance for the alkenic protons observed at δ 7.20. This assignment was further confirmed by both 1H–1H
COSY and 1H–13C correlation experiments which also served to identify the alkenic carbons (δ 132.3). The product was further characterised by a structural study, the results of which appear in Fig. 3 with selected bond lengths and angles in Table 1. The molecule again lies across a crystallographic inversion centre at the mid-point of the C(18)–C(18A) bond, the length of which, 1.334(2) Å, is consistent with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. The C(17)–C(18)–C(18A) angle of 125.7(2)° is consistent with sp2 hybridisation for C(18), and the trans- or E-conformation of the double bond is confirmed by the requirement for a molecular inversion centre.
C double bond. The C(17)–C(18)–C(18A) angle of 125.7(2)° is consistent with sp2 hybridisation for C(18), and the trans- or E-conformation of the double bond is confirmed by the requirement for a molecular inversion centre.
 | ||
| Fig. 3 Molecular structure of alkene 3 showing 50% probability ellipsoids, hydrogen atoms as arbitrary sized spheres. Atoms labelled with the suffix “A” are generated by the symmetry operation (1 − x, −y, −z). | ||
Careful removal of the SiMe2tBu group from 1a and 1b using Bu4NF at low temperature generates nascent (Bu4F)[1-{o-(XCH2C6H4CH2)}-1,2-C2B10H10], which has a nucleophilic site at the carborane 2-carbon atom, which can attack the free benzyl halide. Intramolecular attack results in cyclization to generate dihydronaphthocarborane, whilst it is also possible to envisage intermolecular attack and combinations of inter- and intra-molecular attack leading to polymers and cyclic oligomers respectively. High temperatures need to be avoided for this reaction, since wet Bu4NF is a potent reagent for the deboronation of closo-carboranes under these conditions.24 The spectroscopic properties of the single product of this reaction, 4, are identical to those previously reported for dihydronaphthocarborane,15 although NMR cannot uniquely discriminate between this and cyclic dimers or trimers of the same unit. These oligomers are expected to fragment readily under mass-spectrometry conditions, so that this technique is also not able to uniquely confirm the product formula. For this reason, the molecular structure of 4 was determined by X-ray diffraction and confirms that the product is indeed dihydronaphthocarborane; the structure is shown in Fig. 4 with selected bond lengths and angles in Table 1. The structure can be compared with that of “dihydrobenzocarborane”.25 The eight carbon atoms of the xylyl ring in 4 are essentially planar, with a maximum deviation from the least-square plane of 0.009 Å for C(14). One notable feature is a significant dishing of the molecule, as apparently required by the presence of sp3 carbon atoms at C(11) and C(18), so that the C(1)–C(11)–C(12)–C(17)–C(18)–C(2) ring in 4 is far from planar, the largest deviation from a least-squares plane is 0.18 Å, whilst the analogous ring in dihydrobenzocarborane is planar, with a largest deviation of 0.033 Å.
 | ||
| Fig. 4 Molecular structure of dihydronaphthocarborane 4 showing 50% probability ellipsoids, hydrogen atoms as arbitrary sized spheres. | ||
In conclusion, we have demonstrated that (halo-o-xylyl)-o-carboranes are not suitable precursors to amine functionalised carborane ligands analogous to constrained geometry cyclopentadienyl ligands. Coupling reactions between the halo-o-xylyl units provide a means of preparing precursors to the rational stepwise construction of oligomeric carborane materials.
Experimental
All manipulations of air- and moisture-sensitive compounds were performed on a conventional vacuum/nitrogen line using standard Schlenk and cannula techniques or in a nitrogen filled glove box. When required, solvents were dried by prolonged reflux over the appropriate drying agent, prior to distillation and deoxygenation by freeze–pump–thaw processes where appropriate. NMR solvents were vacuum-distilled from suitable drying agents and stored under a dry nitrogen atmosphere. Elemental analysis was performed by the micro-analytical service within this department on an Exeter Instruments analyser. NMR spectra were recorded on the following instruments: Varian Unity-300 (1H, 11B, 13C), Varian 500 (1H, 13C, HETCOR), 1H and 11B NMR were recorded on the Unity-300 unless otherwise stated. All chemical shifts are reported in δ (ppm) and coupling constants in Hz. 1H NMR spectra were referenced to residual protio impurity in the solvent (CHCl3, 7.26 ppm). 13C NMR spectra were referenced to the solvent resonance (CDCl3, 77.0 ppm). 11B NMR were referenced externally to Et2O·BF3δ = 0.0 ppm. Except where otherwise indicated, all spectra were recorded in CDCl3 at ambient temperature.CAUTION: α,α′-dihalo-o-xylenes and compounds 1a and 1b are potent irritants and lachrymators and as such skin contact should be avoided.
Syntheses
X-Ray crystallography
Single crystal X-ray diffraction experiments were carried out with a SMART 1K CCD area detector, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The reflection intensities for 1b were corrected by means of a ψ-scan, the reflections for the other compounds were not corrected for absorption. The structures were solved by direct methods and refined by full-matrix least squares against F2 of all data, using SHELXTL programs.26 Crystal data and experimental details are listed in Table 2.CCDC reference numbers 168850–168854.
See http://www.rsc.org/suppdata/dt/b1/b107276e/ for crystallographic data in CIF or other electronic format.
| Compound | 1a | 1b | 2 | 3 | 4 |
|---|---|---|---|---|---|
| Empirical formula | C16H33B10ClSi | C16H33B10BrSi | C32H66B20Si2 | C32H64B20Si2 | C10H18B10 |
| Formula weight | 397.06 | 441.52 | 723.23 | 721.21 | 246.34 |
| Temperature/K | 120(2) | 120(2) | 100(2) | 120(2) | 103(2) |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n | P21/n | P21/c |
| a/Å | 7.466(2) | 7.4927(2) | 9.329(1) | 10.731(2) | 7.215(1) |
| α/° | 107.79(3) | 72.0640(10) | |||
| b/Å | 10.056(2) | 10.0475(3) | 13.502(2) | 16.232(2) | 20.620(3) |
| β/° | 91.10(3) | 83.1420(10) | 104.505(5) | 110.64(2) | 90.48(1) |
| c/Å | 16.135(3) | 16.2819(5) | 18.097(2) | 13.294(3) | 9.268(1) |
| γ/° | 99.24(3) | 80.7340(10) | |||
| Volume/Å3 | 1135.5(4) | 1147.78(6) | 2206.8(5) | 2167.0(7) | 1378.8(2) |
| Z | 2 | 2 | 2 | 2 | 4 |
| μ/mm−1 | 0.222 | 1.844 | 0.105 | 0.107 | 0.056 |
| Reflections measured | 14262 | 8297 | 24049 | 22108 | 11211 |
| Unique reflections | 6087 | 5791 | 5471 | 4965 | 3165 |
| R int | 0.0985 | 0.0391 | 0.0538 | 0.0339 | 0.0493 |
| Reflections I > 2σ(I) | 5057 | 4153 | 4187 | 4135 | 2294 |
| R[F2 > 2σ(F2)] | 0.0458 | 0.0462 | 0.0453 | 0.0358 | 0.0459 |
| wR(F2), all data | 0.1321 | 0.1031 | 0.1209 | 0.0950 | 0.1225 |
Acknowledgements
We acknowledge the award of an EPSRC Senior Research Fellowship (J. A. K. H.), and support from the ERDF Centre for 21st Century Materials at the University of Durham (J. M. M.).References
- A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587 CrossRef CAS; J. Chirinos, T. Rajmankina, A. Parada and F. Ciardelli, Macromol. Chem. Phys., 2000, 201, 2581 CrossRef CAS; S. Park, W. J. Wang and S. P. Zhu, Macromol. Chem. Phys., 2000, 201, 2203 CrossRef CAS; D. van Leusen, D. J. Beetstra, B. Hessen and J. H. Teuben, Organometallics, 2000, 19, 4084 CrossRef CAS; K. C. Hultzsch, P. Voth, K. Beckerle, T. P. Spaniol and J. Okuda, Organometallics, 2000, 19, 228 CrossRef CAS; S. Tian, V. M. Arredondo, C. L. Stern and T. J. Marks, Organometallics, 1999, 18, 2568 CrossRef CAS.
- J. Okuda, Comments Inorg. Chem., 1994, 16, 185 Search PubMed; P. Jutzi and U. Siemeling, J. Organomet. Chem., 1995, 500, 175 CrossRef CAS; C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233 Search PubMed; C. Müller, D. Vos and P. Jutzi, J. Organomet. Chem., 2000, 600, 127 CrossRef CAS; P. Jutzi and T. Redeker, Eur. J. Inorg. Chem., 1998, 663 CrossRef CAS; H. Butenschon, Chem. Rev., 2000, 100, 1527 CrossRef CAS; U. Siemeling, Chem. Rev., 2000, 100, 1495 CrossRef CAS.
- A. K. Hughes, A. Meetsma and J. H. Teuben, Organometallics, 1993, 12, 1936 CrossRef CAS; A. K. Hughes, S. M. B. Marsh, J. A. K. Howard and P. S. Ford, J. Organomet. Chem., 1997, 527, 195 CrossRef CAS; A. K. Hughes and A. J. Kingsley, J. Chem. Soc., Dalton Trans., 1997, 4139 RSC.
- R. N. Grimes, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon Press, Oxford, 1995, vol 1, ch. 9 Search PubMed; M. F. Hawthorne, Acc. Chem. Res., 1968, 1, 281 Search PubMed; A. K. Saxena and N. S. Hosmane, Chem. Rev., 1993, 93, 1081 CrossRef CAS.
- A. S. Batsanov, A. E. Goeta, J. A. K. Howard, A. K. Hughes and J. M. Malget, J. Chem. Soc., Dalton Trans., 2001, 1820 RSC.
- K. Vyakaranam, S. Li, C. Zheng and N. S. Hosmane, Inorg. Chem. Commun., 2001, 4, 180 CrossRef CAS.
- S. Gomez, C. Viñas, M. Lamrani, F. Teixidor, R. Kivekäs and R. Sillanpää, Inorg. Chem., 1997, 36, 3565 CrossRef CAS; F. Teixidor, S. Gomez, M. Lamrani, C. Viñas, R. Sillanpää and R. Kivekäs, Organometallics, 1997, 16, 1278 CrossRef CAS.
- G. F. Zi, Q. C. Yang, T. C. W. Mak and Z. W. Xie, Organometallics, 2001, 20, 2359 CrossRef CAS; K. L. Chui, Q. C. Yang, T. C. W. Mak and Z. W. Xie, Organometallics, 2000, 19, 1391 CrossRef CAS; S. W. Wang, Q. C. Yang, T. C. W. Mak and Z. W. Xie, Organometallics, 2000, 19, 334 CrossRef CAS.
- W. Tjarks, J. Organomet. Chem., 2000, 614, 37 CrossRef; A. H. Soloway, W. Tjarks, B. A. Barnum, F. G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515 CrossRef CAS.
- J. March, Advanced Organic Chemistry, 3rd edn., Wiley, Chichester, 1985, p. 380 Search PubMed.
- J. March, Advanced Organic Chemistry, 3rd edn., Wiley, Chichester, 1985, p. 1106 Search PubMed.
- A. K. M. Annisuzzaman and R. L. Whistler, J. Org. Chem., 1972, 37, 1201 CrossRef; A. K. M. Annisuzzaman and R. L. Whistler, Carbohydr. Chem., 1980, 8, 297 Search PubMed.
- J. G. Wilson, A. K. M. Anisuzzaman, F. Alam and A. H. Solway, Inorg. Chem., 1992, 31, 1955 CrossRef CAS.
- A. Varadarajan, R. M. Sharkey, D. M. Doldgenberg and M. F. Hawthorne, Bioconjugate Chem., 1991, 2, 102 CrossRef CAS.
- D. S. Matteson and R. A. Davis, Inorg. Chem., 1974, 13, 859 CrossRef CAS; D. S. Matteson and R. A. Davis, J. Chem. Soc. D, 1970, 669 RSC ; see also A. Z. Bradley, A. J. Link, K. Biswas, D. Kahne, J. Schwartz, M. Jones Jr., Z. Zhu and M. S. Platz, Tetrahedron Lett., 2000, 41, 8691 Search PubMed.
- F. A Gomez, S. E. Johnson and M. F. Hawthorne, J. Am. Chem. Soc., 1991, 113, 5915 CrossRef CAS.
- F. A. Gomez and M. F. Hawthorne, J. Org. Chem., 1992, 57, 1384 CrossRef CAS.
- W. Jiang, I. T. Chizhevsky, M. D. Mortimer, W. Chen, C. B. Knobler, S. E. Johnson, F. A. Gomez and M. F. Hawthorne, Inorg. Chem., 1996, 35, 5417 CrossRef CAS.
- N. Blažević, D. Kolbah, V. Šunjić and F. Kajfež, Synthesis, 1979, 161 CrossRef CAS.
- L. I. Zakharkin and A. V. Grebenikov, Izv. Akad. Nauk SSSR, Ser. Chim., 1966, 2019 Search PubMed; M. F. Hawthorne, D. C. Young, J. Dvorak, H. Smith, N. Schwartz, M. S. Cohen and M. M. Fein, Inorg. Chem., 1963, 2, 1120 CrossRef.
- X. Yang, W. Jiang, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1992, 114, 9719 CrossRef CAS; P. F. H. Schwab, M. D. Levin and J. Michl, Chem. Rev., 1999, 99, 1863 CrossRef CAS.
- For a recent review of this area see P. Kaszynski, Collect. Czech. Chem. Commun., 1999, 64, 895 Search PubMed.
- H. M. Colquhoun, P. L. Herbertson, K. Wade, I. Baxter and D. J. Williams, Macromolecules, 1999, 31, 1694 CrossRef CAS.
- H. Tomita, H. Luu and T. Onak, Inorg. Chem., 1991, 30, 812 CrossRef CAS; M. A. Fox, J. A. H. MacBride and K. Wade, Polyhedron, 1997, 16, 2499 CrossRef CAS; M. A. Fox and K. Wade, J. Organomet. Chem., 1999, 573, 279 CrossRef CAS; J. Yoo, J.-W. Huang and Y. Do, Inorg. Chem., 2001, 40, 568 CrossRef CAS.
- R. C. B. Copley, M. A. Fox, W. R. Gill, J. A. K. Howard, J. A. H. MacBride, R. J. Peace, G. P. Rivers and K. Wade, Chem. Commun., 1996, 2033 RSC.
- SHELXTL-NT, Version 5.1 , Bruker Analytical X-Ray Instruments Inc., Madison, Wisconsin, USA, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: rotatable 3-D crystal structure diagrams in CHIME format. See http://www.rsc.org/suppdata/dt/b1/b107276e/ |
| This journal is © The Royal Society of Chemistry 2002 |