Fullerene–porphyrin architectures; photosynthetic antenna and reaction center models
Dirk M.
Guldi
Radiation Laboratory, University of Notre Dame, IN 46556, USA.. E-mail: guldi.1@nd.edu
First published on 18th December 2001
Abstract
Fullerenes and porphyrins are molecular architectures ideally suited for devising integrated, multicomponent model systems to transmit and process solar energy. Implementation of C60 as a 3-dimensional electron acceptor holds great expectations on account of their small reorganization energy in electron transfer reactions and has exerted a noteworthy impact on the improvement of light-induced charge-separation. This article describes how the specific compositions of porphyrin chromophores linked to C60—yielding artificial light harvesting antenna and reaction center mimics—have been elegantly utilized to tune the electronic couplings between donor and acceptor sites and the total reorganization energy. Specifically, the effects that these parameters have on the rate, yield and lifetime of the energetic charge-separated states are considered.
 Dirk M. Guldi Dirk M. Guldi | Dirk M. Guldi graduated from the University of Cologne (Germany) in 1988, from where he received his PhD in 1990. In 1992, after a postdoctoral appointment at the National Institute of Standards and Technology, he took a research position at the Hahn-Meitner-Institute Berlin. After a brief stay as a Feodor-Lynen Stipend (Alexander von Humboldt Foundation) at Syracuse University he joined in 1995 the faculty of the Notre Dame Radiation Laboratory where he was promoted to Associate Scientist in 1996. In 1999 he completed his Habilitation at the University of Leipzig (Germany). He was awarded the ‘Heisenberg-Prize’ (1999; Deutsche Forschungsgemeinschaft) and ‘Grammaticakis-Neumann-Prize’ (2000; Swiss Society for Photochemistry and Photophysics). His primary research interests are in the areas of charge separation in super- and supramolecular systems with three-dimensional electron acceptors and organized thin films at semiconductor surfaces as tools viable for the efficient conversion of solar energy. |
1 Introduction
In photosynthesis, cascades of short-range energy transfer and electron transfer events occur between well-arranged organic pigments (i.e., light harvesting antenna ensemble and photosynthetic reaction center (PRC)) and other cofactors.1 Thereby the antenna portion captures light and transduces the resulting excitation energy, via singlet–singlet energy transfer, to the PRC. In the PRC charges are then separated with remarkable efficiency to yield a spatially and electronically well-isolated radical pair. Key to this success is, without any doubt, the overall small reorganization energy (λ ∼ 0.2 eV) exhibited by the PRC and the well-balanced electronic coupling between each donor and acceptor. The arrangement of the donor–acceptor couples in the PRC is simple and accomplished via their non-covalent incorporation into well-defined transmembrane proteins. Owing to the importance and complexity of natural photosynthesis, the study thereof necessitates suitable simpler models. The ultimate goal is to design and assemble artificial systems, which can efficiently process solar energy, replicating the natural analog.2The rich and extensive absorptions (i.e., π–π* transitions) seen in porphyrinoid systems—the pigments of life—hold particular promise for increased absorptive cross sections and, thus, an efficient use of the solar spectrum. Over the course of recent years they emanate as light harvesting building blocks in the construction of molecular architectures.3 Their high electronic excitation energy, typically exceeding 2.0 eV, powers a strongly exergonic electron transfer, which subsequently intercedes the conversion between light and chemical/electrical energy.
The search for an electron transfer partner brings us, however, well beyond components found in photosynthesis on earth, namely, C60. In fact, its discovery was attained in conjunction with compositionary issues in the interstellar medium.4 This new three-dimensional electron acceptor is now readily available and exhibits exciting characteristics. The delocalization of charges—electrons or holes—within the giant, spherical carbon framework (diameter >7.5 Å) together with the rigid, confined structure of the aromatic π-sphere offers unique opportunities for stabilizing charged entities. Six equally-spaced reduction waves in electrochemical experiments,5 with the first reduction step resembling that of quinones—the electron acceptor unit in PRC proteins which is reduced to a semiquinone and finally to a hydroquinone—are a first manifestation of conditions that guarantee the optimal delocalization of charges. In other words, even in a highly reduced fullerene state (i.e., tetraanion, pentaanion and hexaanion) electrons, as they are subsequently added to the fullerene’s π-system, experience little, if any, repulsive forces. From this observation we infer the potential of fullerenes to possess quite small reorganization energies in electron transfer reactions, which renders application of this carbon material as electron accepting moieties particularly appealing under aspects of energy conversion and energy storage.
Thanks to the pioneering protocols, regarding the general chemical functionalization of fullerenes,6 virtually any functional group can be covalently linked with any specific regioisomeric pattern to the highly reactive carbon framework en route to synthetic models of light harvesting arrays and reaction centers.
2 Reorganization energy of fullerenes in electron transfer reactions
The total reorganization energy (λ) is composed of a solvent-independent and a solvent-dependent fragment, λv and λs, respectively.7 In C60, the λv contribution, stemming from different nuclear configurations associated with the transformation, for instance, in a photochemical reaction from an initial to a final state, is very small (λv ∼ 0.06 eV).8 A sound interpretation for this striking observation implies the structural similarity between C60 in the ground, reduced and also excited state. This relates primarily to the rigidity of these spherical carbon structures (i.e., high strain energy), preventing major structural or geometrical changes at room temperature. Further support for this conclusion was lent from small Stokes shifts in excitation experiments and small Raman shifts under reductive conditions. It is also believed that the solvent-dependent term is small, thus requiring only little energy for the adjustment of a generated state (e.g., excited or reduced state) to the new solvent environment. This corresponds directly to the symmetrical shape and large size of the fullerene framework.The sum of these effects bears fundamental consequences upon the classical Marcus treatment of electron transfer reactions. In particular, the Marcus theory predicts that the dependence of electron transfer rates on the free energy changes of the reaction (−ΔGET°) is a parabolic curve.7 In the ‘normal region’ of the Marcus curve (−ΔGET° < λ) the theory predicts an increase in rate with increasing thermodynamic driving force until optimal conditions are reached, when the driving force equals the overall reorganization energy (−ΔGET° ∼ λ). Beyond this thermodynamic maximum the highly exergonic region (−ΔGET° > λ) is entered, where the rate constants start to decrease with increasing free energy changes (‘inverted region’). Variation of λ is not only the key to control the maximum of the parabola, but, most importantly, to influence the shape of the underlying dependence. In principle, smaller λ-values assist in reaching the maximum of the Marcus parabola at smaller −ΔGET° values and, in turn, in shifting the energy-wasting charge-recombination deep into the Marcus ‘inverted region’.
Our own studies, focusing on pulse-radiolytic electron transfer dynamics between fullerenes and a series of radiolytically generated arene π-radical cations, (arene)˙+, with varying oxidation potentials were designed to efficaciously exploit their reorganization energy.9 Interestingly, parabolic dependencies of the rate constants on the thermodynamic driving force (−ΔGET°) were found for intermolecular reactions involving the higher fullerenes C76 and C78. It should be noted that these cases represent some of the rare confirmations of the existence of the ‘inverted region’ in a truly intermolecular forward electron transfer. From these experiments an experimental value of ca. 0.6 eV (in dichloromethane) was deduced for the total reorganization energy (λ) of C76
and C78 in oxidative electron transfer processes. A more recent follow-up work, in which the electrochemically determined oxidation potentials are used for the (−ΔGET°) versus (kET) dependence, instead of the ionization potentials, suggests an even smaller value.
| C |
| 76 |
| /C |
| 78 |
| + (arene)˙ |
| + |
| → (C |
| 76 |
| )˙ |
| + |
| /(C |
| 78 |
| )˙ |
| + |
| + arene |
3 Porphyrin–fullerene van der Waals interactions
Another fascinating scenario involves the utilization of strong π–π associations between metalloporphyrins (MP), in general, and fullerenes as a means to engineer supramolecular arrays with remarkable photoactive and magnetic properties.10 This aspect was systematically explored in a series of MP/C60 cocrystallates with M being Mn, Co, Ni, Cu, Zn and Fe. Favorable van der Waals attractions between the curved π-surface of the fullerene and the planar π-surface of MP, assist in the supramolecular recognition, overcoming, however, the necessity of matching a concave-shaped host with a convex-shaped guest structure. This leads to complexes with unusually short contacts (2.7–3.0 Å), shorter than ordinary van der Waals contacts (3.0–3.5 Å), and a variety of crystal structures, ranging from zigzag-chains to columns (Fig. 1). | ||
| Fig. 1 A view of chains of C60·2CoOEP.10a | ||
In conclusion, van der Waals attractions between porphyrins and fullerenes, which are also appreciable in condensed media (vide infra), constitute an important organization principle: whenever affirmed by the molecular topology of the system these moieties spontaneously tend to achieve close spatial proximity relative to each other.
4 Intermolecular electron transfer reactions
The first ZnP/C60 system probed was part of a larger investigation, focusing on intermolecular electron transfer reactions between a series of radiolytically generated one-electron reduced metalloporphyrins and C60:9a| ZnP˙− + C60 → ZnP + C60˙− |
Interestingly, the rates hardly differ in defiance of a large variation in driving force for the examined metalloporphyrins (−ΔGET° = 0.2–0.8 eV), a result typical for nearly diffusion-controlled reactions. In the toluene–acetone–propan-2-ol solvent composite (8 ∶ 1 ∶ 1 v/v) kdiff amounts to 3.5 × 109 M−1s−1. Despite these facts, the low reduction potentials of tin(IV) porphyrins (SnP) led to the observation of equilibrium conditions between these two molecules (SnP and C60) and their respective one-electron reduced forms (SnP˙− and C60˙−). Rate and equilibrium constants have been established by following the decay and formation kinetics, of the SnP˙− and C60˙−,
respectively, as well as by determination of their relative yields at different [C60]/[SnP] concentration ratios. The actual figures are: k = 3.2 × 109 M−1s−1, k−
= 2.1 × 108 M−1s−1, and K = 14 ± 3. The equilibrium constant was used to calculate an even smaller λ-value for C60 in electron transfer of 0.48 eV. The smaller value may be, in part, due to the intrinsic properties of the solvent mixtures (i.e., relative permittivity and refractive index).
SnP˙− + C60
![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) SnP + C60˙− SnP + C60˙− |
It should be stated that the radiolysis experiments led to just one radical species—either a one-electron reduced MP˙− or C60˙−—without, however, producing the corresponding counter radical cation. By contrast, during the course of the photolysis, ion pairs, that is the radical anion and radical cation, are formed. In polar solvents, for instance, an intermolecular electron transfer evolves in ZnP/C60 mixtures from both triplet states, 3*ZnP (P denotes tetraphenyl- and octaethylporphyrin) and 3*C60 to yield ZnP˙+/C60˙− radical pairs. The exact pathway depends on the excitation wavelength, exciting either ZnP or C60:11
| 3*ZnP + C60 → ZnP˙+ + C60˙− |
| ZnP + 3*C60 → ZnP˙+ + C60˙− |
Spectroscopically, the photoproducts are identified with relative ease by characteristic absorption changes in the visible and near-infrared: maxima around 650 nm are clear attributes of the ZnP˙+, while transient absorbances at 1000 nm resemble the diagnostic marker of the fullerene radical anion, C60˙−.
The quantum efficiency for electron transfer is higher upon employing the better electron donating octaethylporphyrin in combination with C60 than the tetraphenylporphyrin analog. Conversely, non-polar media favor intermolecular triplet–triplet energy transfer from 3*ZnP to C60:
| 3*ZnP + C60 → ZnP + 3*C60 |
Decisive evidence for such electron transfer processes—via the triplet–triplet route—came from FT-EPR experiments,11b,c which give rise to resonance peaks associated with C60˙− and 3*C60, corroborating that both energy and electron transfer processes occur between ZnP and C60. Furthermore, from the analyses of the spin polarization and the time-evolution, it was concluded that the electron spin polarization of 3*ZnP is transferred to 3*C60, conserving the spin angular momentum.
5 Stabilization of the charge-separated state—charge-recombination in the inverted region
Considering the experimentally determined λ values of 0.48 and 0.6 eV,9 which are remarkably small compared to other artificial model acceptors (0.8–1.2 eV), the thermodynamic maximum (−ΔGET° = λ) and, therefore, access to the ‘inverted-region’ (−ΔGET° > λ) should be reached with relative ease. Now, any reaction that renders strongly exothermic shifts deep into the ‘inverted-region’, as charge-recombination processes typically are, and its rate is largely slowed-down. An additional benefit of a small λ value is that the ‘normal-region’ (−ΔGET° > λ) becomes steeper, which is expected to accelerate the charge-separation.In this regard, a finding by Gust et al. is of fundamental importance.12 In the first example of a C60-based system (1), in which the two π-electron systems are essentially in van der Waals contact, they found that charge-recombination is significantly slower than charge-separation. This pioneering work evoked the synthesis of a virtually unlimited number of C60-based donor–acceptor ensembles integrating a variety of organic electron-donor molecules via covalent bonds.12–16

A particularly promising set of ensembles turned out to be the choice of ZnP, covalently linked, for instance, to several pyrrolidino- or methanofullerene derivatives as artificial reaction centers, in which the complicated natural mechanism can be reduced to its basic elements. An exemplification is given in Fig. 2. Excitation of the ZnP portion with visible light, which leads predominantly to the population of its first singlet excited state, 1*ZnP, is followed by a rapid intramolecular electron transfer (kCS) to yield a long-lived charge-separated state in high yields:

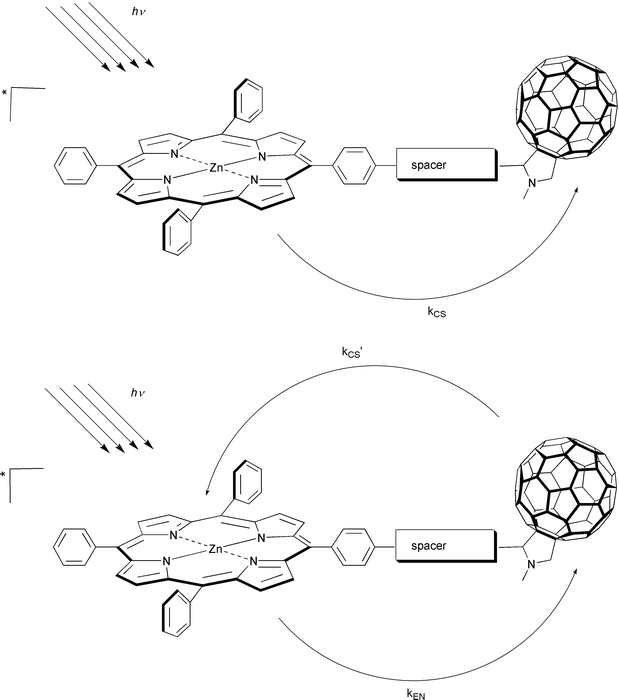 | ||
| Fig. 2 Schematic illustrations of the charge-separation pathways in ZnP-C60 ensembles, following visible light excitation. | ||
Importantly, a significant fraction of the photon energy is converted and stored in the form of ZnP˙+–C60˙−. Alternatively, the energetic charge-separated state may evolve from 1*C60 (kCS′). This pathway is, however, of minor importance, since it infers either the very unlikely event of direct C60 excitation or the competing transfer of singlet excitation energy (kEN).
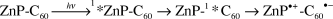
To exemplify the advantages of incorporating a fullerene, a C60-based porphyrin dyad should be compared with a quinone-based analog.17 Since comparable reduction potentials, donor–acceptor separations and electronic couplings guarantee similar −ΔGET°’s, any effects must be connected with the different reorganization energies. In fact, accelerated charge-separation (∼6 times) and decelerated charge-recombination processes (∼25 times), by which the C60-based system excels the corresponding quinone dyad, are an impressive proof of the above hypothesis.
Thus, at the beginning of our own photophysical investigation, we anticipated seeing a deceleration of the energy-wasting and undesirable charge-recombination. Recently we accomplished a definite verification of this assumption by demonstrating that charge-recombination, in a series of C60-containing donor–acceptor ensembles, occurs indeed in the ‘inverted-region’.18 Here, the selection of ZnP showed how crucial is the correct choice of the intramolecular separation in light-driven electron transfer reactions, especially to ensure −ΔG° >> λ. A novel fullerene–porphyrin conjugate with van der Waals contacts (edge-to-edge separation (Ree) ∼3.0 Å) such as, for example trans-2-ZnP-C60, reminiscent of the trans-1-ZnP-C60 ensemble (not shown) reported by Diederich et al.,13h provided an exquisite setting for this study. The short separation guaranteed that an intramolecular charge-separation succeeds in virtually any solvent and dominates over the competing energy transfer. This is illustrated in Fig. 3, showing the rapid formation and decay of ZnP˙+ between 670–680 nm and C60˙− around 900 nm in toluene, due to charge-separation and charge-recombination processes, respectively. Changing the solvent polarity between non-polar toluene and polar benzonitrile provided the means to alter the free energy changes over a wide range. But, most importantly, a marked acceleration of the charge-recombination rates was seen at smaller −ΔGCR°, namely, at higher relative permittivities, which corroborated nicely our working hypothesis. For instance, the lifetimes varied between 619 ps (toluene) and 38 ps (benzonitrile). Correlating log (kET) with −ΔGET° (i.e., charge-separation and charge-recombination) and fitting of the resulting parabolic dependence yielded an experimental λ value of 0.86 eV. A possible explanation for this somewhat large value (vide infra) is that the tight stacking of the two moieties is a responsible force for mutually perturbing the π-systems.
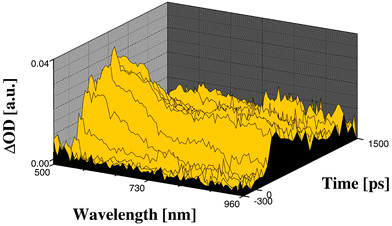 | ||
| Fig. 3 Differential absorption spectra obtained upon picosecond flash photolysis (532 nm) of ∼10−5 M solutions of trans-2-ZnP-C60 in nitrogen saturated toluene with time delays between −300 and 1500 ps, showing the growth and decay of the charge-separated state. | ||
An independent and elegant work by Schuster et al. manifested that the same outstanding trend holds also in a parachute-ZnP-C60:19 slower charge-recombination dynamics at larger −ΔGCR°. However, due to the slightly looser stacking, a longer-lived ZnP˙+-C60˙− radical pair (69 ps in benzonitrile) was seen than in trans-2-ZnP-C60. A recent modeling suggests that the porphyrin bends over to sit in a much closer position, relative to the fullerene, than earlier anticipated. The tendency, that a wider separation retards the charge-recombination, points already to an important direction (vide infra).
In a different approach, the charge-recombination rates in pyrrole-ZnP˙+-C60˙− with pyrrole-H2P˙+-C60˙− (H2P = free base tetraphenylporphyrin) were identified as a potent means for varying the free energy changes (−ΔGCR°), namely, from 1.38 eV to 1.58 eV without, however, modifying the medium (i.e., benzonitrile).20 The corresponding lifetimes of 290 ps and 50 ps furnish essentially the same conclusion, viz. the dynamics within these radical pairs are consistent with the occurrence of charge-recombination in the ‘inverted region’ of the Marcus parabola.
6 Tuning the coupling, orientation and separation in donor–acceptor dyads
In this review, the discussion shall be limited to some selected examples outlining several unprecedented trends, although an armada of ZnP-C60 arrays has appeared in recent years.12,13,15–20 The examples were chosen to disclose the most fundamental and most far-reaching impact that fullerene research imposes on our understanding of charge-separation processes. Here the overriding principles can be summarized as gaining control over the (i) electronic coupling, (ii) geometrical overlap and (iii) nature of the intervening spacer in donor–acceptor ensembles. The key objective is to highlight parameters associated with the formation of energetic charge-separated states in synthetic reaction center models.At this point, reference should be made to the excellent contributions by Imahori and Sakata15a,d and Gust, Moore and Moore,15h,i in which several key issues were reviewed that are, however, not covered in the present work, such as synthetic aspects.
In general, attachment of the various porphyrin moieties—porphyrin malonates or porphyrin aldehydes—to C60 has been achieved by standard reactions in fullerene chemistry, such as cyclopropanation reactions (so called Bingel reaction),6 1,3-dipolar cycloaddition of azomethine ylides,21a or else [4 + 2] cycloadditions.21b These reactions imply relatively simple steps and have afforded a wide variety of fullerene–porphyrin ensembles.12,13,15–20
The first class encompasses linear arrays, in which a systematic variation of the spatial distance, separating the donor (ZnP) from the acceptor (C60), to about 11.9 Å (Ree), but preserving the overall free energy changes (i.e., in polar solvents), leads to lifetimes as large as 2.7 microseconds in deoxygenated THF (see Table 1).22 The reader should be reminded of the picosecond lifetime in the closest ZnP/C60 packing possible (see trans-2-ZnP-C60). Thus, this approach has increased the lifetime of charge-separation by a factor of nearly 7000. All these model systems are structurally well-defined assemblies in which both moieties are held at fixed distances and orientations. The reorganization energies, as far as they were determined, describe an interesting distance dependence: first they drop from 0.86 eV (Ree = 3.0 Å) to ∼0.5 eV (Ree = 6.18 Å)18 before they steadily increase to 0.66 eV (Ree = 11.9 Å).21 On the other hand, the electronic coupling (V) decreases with distance throughout all the systems from 415 cm−1 (Ree = 3.0 Å) to 3.9 cm−1 (Ree = 11.9 Å). Therefore, in general, it can be stated that the reorganization energies are indeed smaller than those reported previously for porphyrin–quinone and zinc porphyrin–free base porphyrin linked systems, which typically are in the range between 0.8 eV and 1.2 eV.23
| Compound | THF | Benzonitrile | DMF | Other solvents |
|---|---|---|---|---|
| a Frozen matrix. b Toluene. c 2-Methyltetrahydrofuran. d o-Dichlorobenzene. | ||||
| trans -2-ZnP-C 60 | 385 ps | 38 ps | 619 psb | |
| parachute -ZnP-C 60 | 99 ps | 69 ps | 56 ps | |
| parachute -H 2 P-C 60 | 314 ps | 155 ps | 107 ps | |
| pyrrole -ZnP-C 60 | 50 ps | |||
| pyrrole -H 2 P-C 60 | 290 ps | |||
| equatorial -ZnP-C 60 | 2.6 μs | 1.1 μs | 0.21 μs | |
| meta -ZnP-C 60 | 215 ns | 113 ns | 99 ns | |
| para -ZnP-C 60 | 236 ns | 149 ns | 133 ns | |
| norbornylogous -ZnP-C 60 | 420 ns | |||
| amide -ZnP-C 60 | 2.7 μs | 0.78 μs | 0.57 μs | |
| ZnP-H 2 P-C 60 | 34 μs | 21 μs | 20 μs | |
| Fc-ZnP-C 60 | 3.7 μs | 7.5 μs | 16 μs | |
| Fc-H 2 P-C 60 | 8.3 μs | 19 μs | ||
| Fc-ZnP-H 2 P-C 60 | 0.38 sa | |||
| (ZnP) 3 -ZnP-H 2 P-C 60 | 1.3 nsc | |||
| ZnP- pyridine -C 60 | intermolecular | intermolecular | ||
| DABCO-ZnP-C 60 | 1.98 nsb | |||
| C 60 -ZnP-DABCO-ZnP-C 60 | 2.28 nsb | |||
| meta -ZnP-C 60 -ZnP | 150 nsd | |||
| para -ZnP-C 60 -ZnP | 290 nsd | |||
| para -(DABCO)-ZnP-C 60 -ZnP | 702 nsd | |||
Comparing trans-2-ZnP-C60 with the topographically different equatorial-ZnP-C60, provided a unique set of models suitable for unraveling effects associated with the geometrical arrangement of donor and acceptor (i.e. face-to-face versus face-to-edge).24 The modification of the relative alignment, as it was accomplished synthetically through a different positioning of the two connecting tethers at the fullerene core, alters the π–π interactions. The variance in spatial overlap is reflected in the electronic coupling, which differs by as much as 350 cm−1. Another important result is the change of the charge-separated state lifetime: 4 orders of magnitude difference shifts the lifetime from the lower picosecond (trans-2-ZnP-C60) to the microseconds regime (equatorial-ZnP-C60; benzonitrile: 1.1 μs). Equally important is to realize the fact that the face-to-edge alignment in the equatorial isomer renders a good access to both cores. This affects crucially the solvation stabilization, ensuring much better interactions with the charge-separated radical pair, compared to the less accessible moieties given by the face-to-face geometry in the trans-2 analog, in which, for example, most of the porphyrin surface is covered by the fullerene.
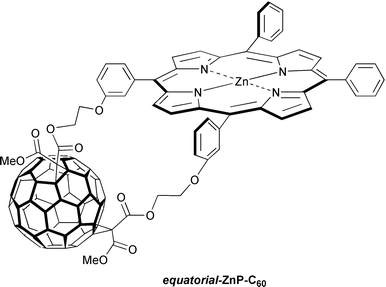
A surprising result comes from a fundamentally different type of linkage, in which flexibility of the spacer unit is being provided by connecting the ZnP/C60 couple with –O-CH2-CH2-O– units of differing composition.24b This contrasts with the work on steroid-,13f amide-,22 norbornylogous-linkages,13b where the structural rigidity of the spacer is the overriding principle. Furthermore, an altered substitution pattern on the ZnP’s phenyl moieties, namely, meta- versus para-, affects the spatial overlap between the two moieties. This notion is further furnished by molecular modeling, which suggests that not only a different Ree, but, most importantly, a deviant overlap governs the meta- and para-ZnP-C60 configuration, as depicted in Fig. 4. Charge-transfer absorption and charge-transfer emission, as meaningful attributes for electronic interactions, indeed corroborate this hypothesis.
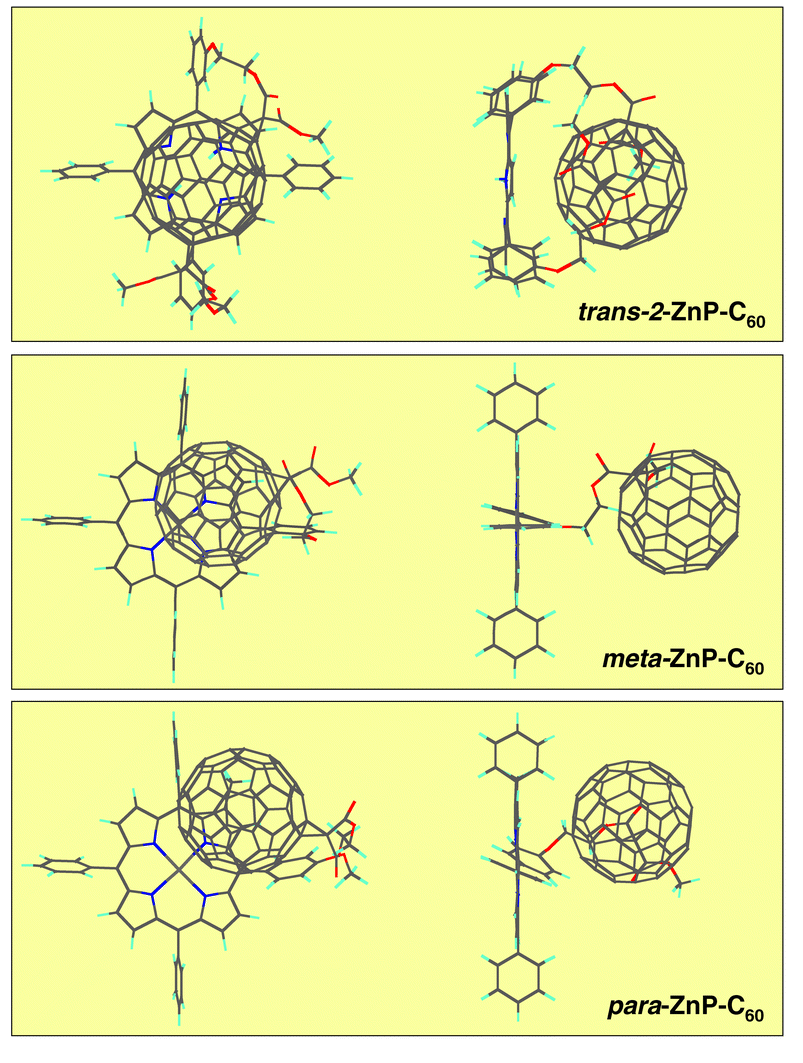 | ||
| Fig. 4 Top and side views of overlapping fullerene and porphyrin moieties in trans-2-ZnP-C60, meta-ZnP-C60 and para-ZnP-C60. | ||
The van der Waals stacked dyad (i.e., trans-2-ZnP-C60; 3 Å) and amide-ZnP-C60 (Ree = 11.9 Å)22 emerge as ideal model systems, due to their limited interaction distance. As far as the charge-transfer absorption in these systems is concerned, its intensity drops as a function of separation in virtually any given solvent in the following order: trans-2-ZnP-C60 (ε = 1470 M−1 cm−1) > meta-ZnP-C60 (ε = 740 M−1 cm−1) > para-ZnP-C60 (ε = 10 M−1 cm−1). By contrast, due to the linear positioning of the redox moieties at opposite ends in amide-ZnP-C60 no charge-transfer features were seen at all.22d Overall, the electronic coupling, which was derived from these spectroscopic assets, reveals a resembling tendency varying from 436 cm−1 to 3.9 cm−1.

The energetic separation between charge-transfer absorption and emission assists in determining the reorganization energy in at least non-polar solvents, such as benzene and toluene. Polar solvents, on the other hand, prevent this type of analysis, since non-radiative processes deactivate the charge-transfer state, instead of the emissive transition. The λ-values are unprecedentedly small, at least in comparison with other artificial model systems and are approaching those established for the PRC (∼0.2 eV). For example, at room temperature meta-ZnP-C60 gives rise to 0.16 eV in toluene. Substantially larger values must, however, be extrapolated for more polar solvents.
In general, all dyads investigated reveal destabilization of the charge-separated state in polar solvents. This trend even holds for para-ZnP-C60, in which the weakest coupling among the series of dyads suggests that the semi-flexible –O-CH2-CH2-O– chain must be the inception for some configurational changes, once photoexcited. Just for comparison, the meta-linkage ensures a better, tighter stacking of the ZnP onto the fullerene moiety (see Fig. 4). Nevertheless, the charge-recombination dynamics are similar in these two flexibly linked donor–acceptor ensembles, namely, several hundred nanoseconds (para-ZnP-C60: 149 ns; meta-ZnP-C60: 113 ns; both in benzonitrile). The small, but notable, changes found in virtually all the investigated solvents, can be correlated with the different donor strength of the meta- versus para-substituted ZnP. As a consequence of the para-isomer’s higher oxidation potential, larger free energy changes determine the charge-recombination process, shifting the kinetics deeper into the inverted region. From this we infer that in the charge-separated state the ZnP˙+ and C60˙− moieties take similar separations relative to each other in both dyads. It cannot be ruled out, however, that the smaller coupling, which prevails in this donor–acceptor ensemble, may have an equal contribution.
One of the major conclusions, as it certainly evolves from all these versatile studies, is that an ideal electron transfer scenario implies charge-separation close to the thermodynamic maximum (−ΔGCS° = λ) to minimize the activation barrier. Based on the data known so far −ΔGCS° ∼0.6 eV appears to be a good reference point. In addition, −ΔGCR° should be as large as possible, namely, >> λ. ZnP-C60 ensembles certainly meet these requirements quite well, which lends important incentives for the strategy of more complex triad and tetrad structures (vide infra). A different aspect of this thermodynamic reasoning reaches out to some technological concerns. It permits the possibility to minimize the loss of excited state energy and, thereby, to improve the conversion efficiency of solar energy into electrical and chemical energy.
In principle, similar effects can be summarized for several H2P-C60 ensembles, making use of flexible and rigid spacer units.13c,f,19,20 An important design consideration implies that the higher oxidation potential of H2P˙+ /H2P couple relative to that of ZnP˙+/ZnP of around 200 mV, would allow storing a larger fraction of the excited state energy as chemical potential in the charge-separated state. Fig. 5 structures the kinetic schemes for the photoinduced relaxation and deactivation processes in H2P-C60 as compared to ZnP-C60. From the energetic point of view, slower kCS and kCR are logical consequences that stem from the energy gap variation in H2P-C60versus ZnP-C60 ensembles.
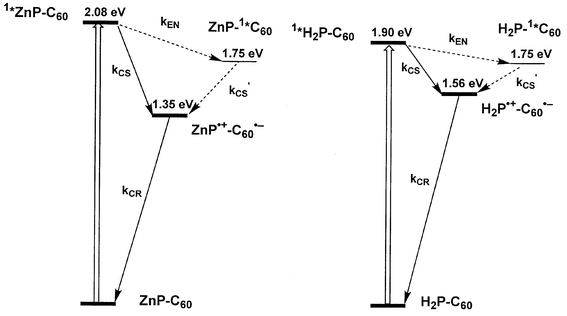 | ||
| Fig. 5 Energetic diagrams illustrating the major photophysical events in ZnP-C60 and H2P-C60 ensembles and the energies of the associated states (i.e., excited states and charge-separated states) following the exclusive excitation of the porphyrin chromophore. | ||
Interestingly, substitution of the electron acceptor unit C60 by C70, which is a slightly better electron acceptor, leads to a two-fold expedition of the charge-separation dynamics (kCS).13e
7 Linear donor–acceptor ensembles—triads and tetrads
Encouraged by these remarkable results, several linear triads (Ree = 30.3 Å) and tetrads (Ree = 48.9 Å) were built around the ZnP/C60 couple as artificial reaction centers. We probed in collaboration with Imahori et al. the lifetime of the charge-separated state via systematically extending the donor–acceptor composition.22,25,26 Despite the multifaceted difficulties, connected with the design of rigidly spaced ensembles containing more than just a single donor–acceptor couple, it is imperative to realize that a long-distance and long-lived charge-separated state can only be attained in a series of short-range, fast and efficient electron transfer systems unless wire-like spacer units are employed. A specific challenge involves attaining a fine-tuned and directed redox gradient along donor–acceptor linked arrays.The first promising results stem from a set of molecular triads in which a fullerene moiety is linked either to an array of two porphyrins (i.e., ZnP and H2P; ZnP-H2P)22a,c or to a Fc-ZnP fragment.25b,d In the ZnP-H2P-C60 triad, the ZnP moiety performs as an antenna molecule, transferring its singlet excited state energy to the lower lying H2P. In polar benzonitrile, this energy transfer is followed by a sequential electron transfer yielding ZnP-H2P˙+-C60˙− (τ = 77 ns) and subsequently ZnP˙+-H2P-C60˙− (τ = 21 μs). Considering the overall efficiency of 40% for (i) funneling light from the antenna chromophore (i.e., ZnP) to the H2P chromophore, (ii) charge-injection into the fullerene core and (iii) charge-shift, this artificial reaction center reproduces the natural system very well.19a

The lifetimes of ZnP˙+-H2P-C60˙− in different media correlate well with the polarity: 34 μs (THF), 21 μs (benzonitrile), 20 μs (DMF). Since the driving force of charge-recombination (−ΔGCR°) decreases even in ZnP-H2P-C60 with increasing solvent polarity, the observed trend suggests that the associated rate constants are in the ‘inverted region’ of the Marcus curve.22c
The function of the Fc-ZnP-C60 and Fc-H2P-C60 systems, on the other hand, is a lot simpler and limited to two consecutive electron transfers yielding the Fc+-ZnP-C60˙− and Fc+-H2P-C60˙− in nearly 82 and 25% yield, respectively.22c,25b In oxygen-free benzonitrile, the final charge-separated state decays, however, considerably faster to the singlet ground state than does the ZnP˙+-H2P-C60˙− (4.8 × 104 s−1). The actual rate constants are 1.3 × 105 s−1 (Fc+-ZnP-C60˙−) and 1.2 × 105 s−1 (Fc+-H2P-C60˙− ). Taking into account the similarity in molecular structure and separation (Ree = 30.3 Å) the different thermodynamics must be responsible for this intrinsic behavior. In fact, variation of the −ΔGCR° (i.e., comparing THF and DMF) led to a surprising discovery: the dynamics within the Fc+/C60˙− couples are in the ‘normal region’ of the Marcus curve, that is, −ΔG < λ.22c

In general, electronic coupling (V) and reorganization energy (λ) further corroborate the trend seen in the ZnP-C60 dyads: diminished couplings and increased reorganization energies accompany the larger separation (Ree = 30.3 Å). Precisely, the following values were derived for the ZnP-H2P-C60/Fc-H2P-C60 ensembles (λ = 1.09 eV; V = 0.019 cm−1) from Fig. 6.
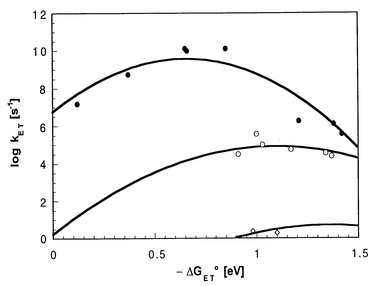 | ||
| Fig. 6 Driving force (−ΔGET°) dependence of intramolecular charge-separation and charge-recombination rate constants in amide-ZnP-C60 dyad (top curve), ZnP-H2P-C60/Fc-H2P-C60 triads (center curve) and Fc-ZnP-H2P-C60 tetrad (bottom curve). | ||
Replacement of Fc by a carotenoid secondary electron donor (Car) in Car-ZnP-C60 and Car-H2P-C60, led to a fundamentally different picture.27 Now, the carotenoid triplet excited state moves energetically between those of the final radical pair and of the ground state. Thus, the product of the charge-recombination is predominantly the 3*Car-H2P-C60, and the dynamics must, although located still in the ‘inverted region’, occur with −ΔGCR° ∼ λ, that is, near the top of the Marcus parabola. For example, in polar solvents −ΔGCR° is 0.6 eV. Most interestingly, lowering the temperature to 77 K increases the lifetime of Car˙+-H2P-C60˙− from 60 ns to 325 ns in 2-methyltetrahydrofuran.
Successful mimicry of the primary events in photosynthesis using ZnP-H2P-C60 and Fc-ZnP-C60 encouraged us to combine these two systems into an integrated single system, Fc-ZnP-H2P-C60 .26 Indeed, the lifetime of the spatially-separated (Ree = 48.9 Å) radical pair, the product of a sequence of energy and multistep electron transfer reactions, reaches well beyond milliseconds (0.38 s), into a time domain which has never been accomplished so far in an artificial photosynthetic reaction center. As reference points 340 μs and 12.7 ms are reported for synthetic systems in room temperature solutions and at 77 K, respectively, which further document the conceptional breakthrough in our work. The lifetime is also comparable, for example, to the lifetimes (∼1 s−1) of the bacteriochlorophyll dimer radical cation ((Bchl)2˙+)–secondary quinone radical anion (QB˙−) ion pair in the bacteria PRC. The relatively low quantum yields (0.17–0.24) can be rationalized by the competition of the various charge-shift reactions, transferring the charge from Fc-ZnP-H2P˙+ -C60˙− to Fc+-ZnP-H2P-C60˙− versus the intrinsic decays of the reactive intermediates. It should also be emphasized that such an extremely long lifetime of the tetrad system has been well correlated with the charge-separated lifetimes of two homologous series of porphyrin–fullerene dyad and triad systems. In essence the longer lifetimes, smaller coupling elements (Fc-ZnP-H2P-C60: 1.7 × 10−4 cm−1) and larger reorganization energies (Fc-ZnP-H2P-C60: 1.32 eV) are clear characteristics of these systems (vide supra).

In Fc-ZnP-H2P-C60, one of the limiting parameters is certainly the charge-injection from 1*H2P into the fullerene acceptor to yield Fc-ZnP-H2P˙+ -C60˙− . To facilitate this crucial step—by avoiding this bottle-neck—the first charge-separation step should be probed in a Fc-ZnP-ZnP-C60 tetrad. In general, ZnP has a lower oxidation potential, thus providing more thermodynamic driving force, and therefore potentially larger rate constants and quantum yields for the crucial conversion of the Fc-ZnP-1*ZnP-C60 intermediate to the Fc-ZnP-ZnP˙+-C60˙− species.
Unquestionably, the most striking and far reaching observation is that charge-recombination in all these ZnP˙+/C60˙− couples (i.e., dyads and triads) is located in the ‘inverted region’ of the Marcus parabola, regardless of linkage, distance and orientation. By contrast, lowering the driving force via replacing the ZnP with the better electron acceptor ferrocene (Fc), while keeping all other parameters (i.e., distance, acceptor, solvent, temperature, etc.) constant, shifts the dynamics into the normal region. This variation is of great advantage in determining parameters such as electronic coupling (V), reorganization energy (λ) and damping factor (β) with high accuracy. They all have key features for material design considerations with the objective being to prolong the lifetime of the energetic charge-separated state, while, simultaneously, optimizing the efficiency of charge separation.
8 Branched donor–acceptor ensembles—hexad
Quite remarkably, a sophisticated (ZnP)3-ZnP-H2P-C60 hexad, comprised of a (ZnP)3-ZnP model antenna system linked to a H2P-C60 reaction center, has been shown to reveal indeed a cascade of efficient light-driven energy and electron transfer processes.28 Specifically, the antenna portion reveals rapid transduction (∼50 ps) of the singlet excited energy from the peripheral (ZnP)3 to the central ZnP, which itself transfers the excitation energy further to the H2P chromophore of the reaction center with a rate of 4.2 × 109 s−1. In the final part of the sequence, the resulting (ZnP)3-ZnP-1*H2P-C60 is the precursor state for an electron transfer process to the electron accepting C60. The net result of this complex chain of events is a 1.3 ns long lived charge-separated (ZnP)3-ZnP-H2P˙+-C60˙− radical pair, formed in 70% yield. In conclusion this hexad mimics the basic function of both natural photosynthetic antenna systems and reaction center complexes.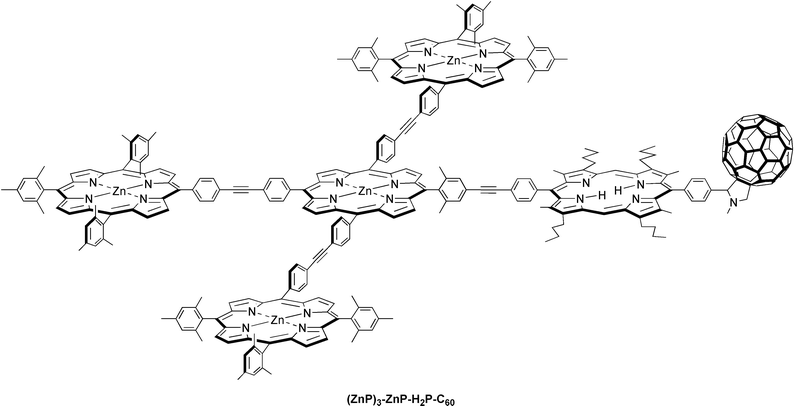
9 Self-assembled donor–acceptor ensembles
A better control over the separation, angular relationships, electronic coupling and composition in donor–acceptor assemblies at a molecular level is a formidable task—especially in artificial antenna and reaction centers—to control the rates and yields of energy and electron transfer reactions and to eliminate the energy wasting charge-recombination. Meaningful incentives can be borrowed from the organization-principle in the bacterial photosynthetic reaction center: the different light- and redox-active components are embedded via non-covalent interactions into a protein matrix. In principle, biomimetic methodologies, such as hydrogen-bonding, donor–acceptor complexation, electrostatic interactions and π–π stacking, guarantee the control over modulating the composition and, simultaneously, achieving well-defined and rigid architectures, with high directionality and selectivity. Thus, self-assembled donor–acceptor ensembles are a viable alternative to supermolecular polyads (e.g., triads, tetrads, etc.), involving covalent links between the components, for increasing the rate, yield and lifetime of the charge-separated state. Typical examples of self-organizing ensembles are illustrated in Fig. 7.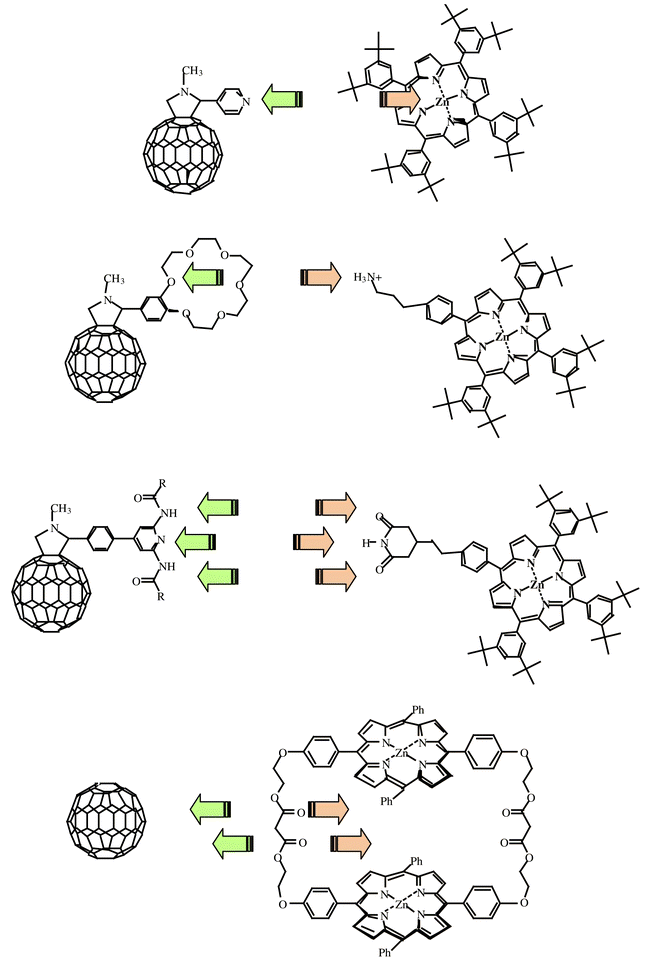 | ||
| Fig. 7 Self-organizing motifs. | ||
A first demonstration was presented in the form of a ZnP-pyridine-C60 complex. In this, the reversible coordination of a pyridine functionalized fullerene ligand (pyridine-C60) to the square-planar zinc center constitutes a labile but, nevertheless, measurable (K
∼ 5000 M−1) binding motif, explored by three different research teams simultaneously.29–31 The ground state features of ZnP in the visible (Q-bands) were employed as sensitive aids to monitor the progression of the ZnP-pyridine-C60 complexation: red-shifted transitions and the observance of clear isosbestic points. In a chain of events (Fig. 8)—triggered by light—the excited donor activates a rapidly occurring electron transfer to the electron accepting C60
within the ZnP-pyridine-C60 complex.29 The weak equilibrium between dissociation and association of the ‘metal–pyridine’ bond facilitates then, in the final step of the sequence, the crucial break-up of the radical pair, before the competing charge-recombination starts to become a restriction. In ZnP-pyridine-C60 the free radical ions (ZnP˙+/C60˙−) live for tens of microseconds in THF and benzonitrile, and any processes, as they may take place, are exclusively governed by an intermolecular diffusion. By contrast, in covalently linked donor–acceptor dyads fast charge-recombination prevails, limiting the lifetime of the radical pair to a few nanoseconds (vide supra).
| ZnP + pyridine-C60
ZnP-pyridine-C60 |
| RuP + pyridine-C60 → RuP-pyridine-C60 |
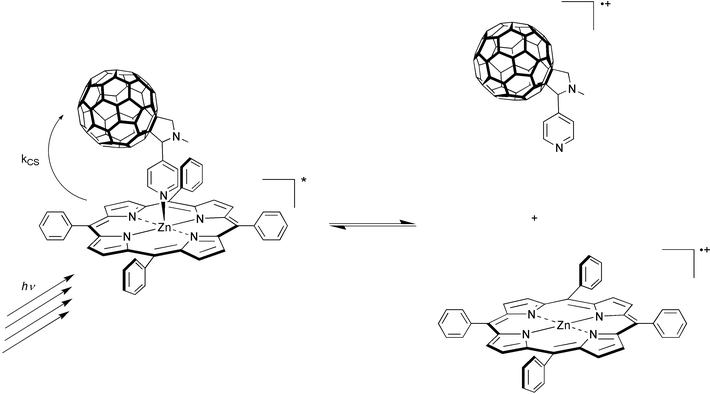 | ||
| Fig. 8 Schematic illustration of the break-up of the ZnP˙+-pyridine-C60˙− radical pair. | ||
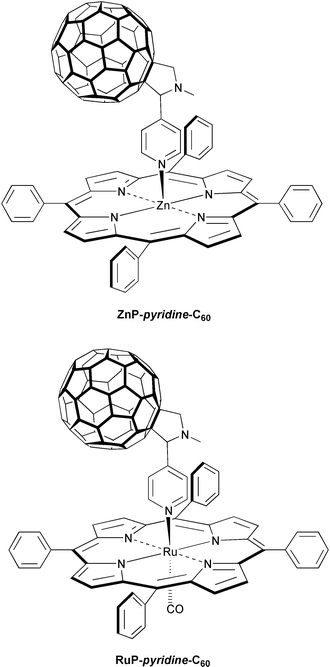
In this regard it appeared attractive to us that complexation of pyridine-C60 to a ruthenium tetraphenylporphyrin (RuP) produces the quite stable RuP-pyridine-C60 complex.29 Here the overriding principle is that utilization of the strong π-back-bonding strengthens the ‘metal–pyridine’ bond relative to that found in the ZnP-pyridine-C60 analog, in which the bonding is limited to a weak σ-character. As a consequence, the intramolecular charge-separated RuP˙+-pyridine-C60˙−, as observed in polar solvents, recombines rapidly on the picosecond time scale (<4000 ps), since the diffusional splitting of the radical pair is largely suppressed.
With the objective to devise linear architectures of higher complexity, that is, triads, tetrads and pentads (vide infra), several ZnP/C60 containing ensembles were subjected to complexation assays with diazabicyclooctane (DABCO).32 The bidentate DABCO ligand exhibits a number of appealing features: it forms not only square pyramidal 1∶1- or 2∶1-complexes with, for example, ZnP, but is also a good electron donor. A prerequisite for their successful construction restricts the use to non-coordinating solvents. Suitable polar media, on the other hand, set up a competition between DABCO and solvent complexation and lead subsequently to dissociation into the free components. Importantly, charge-recombination kinetics in the primary building block (i.e., trans-2-ZnP-C60, etc.)18 of these complexes reveal that the large −ΔGCR° values in toluene are extremely helpful to stabilize the ZnP˙+-C60˙− radical pair. Considering these facts in concert, it is clear that among the many unique fullerene features the small reorganization energy guarantees appreciable affects in these photoactive architectures, especially in light of retarding charge-recombination.
Depending on the relative concentrations, namely, that of DABCO and trans-2-ZnP-C60, the precursor dyad was consecutively transformed into the DABCO-ZnP-C60 triad and the C60-ZnP-DABCO-ZnP-C60 pentad. In these new ensembles, similar electron transfer rates (∼1011 s−1) convert the photoexcited chromophore state into the different charge-separated states. The lifetimes of the latter vary markedly with the ensemble constitution, indicating that subsequent charge-shift reactions, indeed, take place, prior to the decay to the singlet ground state. For example, a significant improvement is seen upon going from trans-2-ZnP˙+-C60˙− (toluene: τ = 619 ps) and DABCO˙+-ZnP-C60˙− (toluene: τ = 1980 ps) to C60-ZnP-DABCO˙+-ZnP-C60˙− (toluene: τ = 2280 ps). The close energy of the DABCO-ZnP˙+-C60˙− and DABCO˙+-ZnP-C60˙− states implies that the positive charge may be distributed over both moieties, instead of being located exclusively on DABCO. In summary, the simple addition of extra components, which self-assemble to the precursor in a controlled manner, is an effective mode to gain control over the charge-recombination rates.
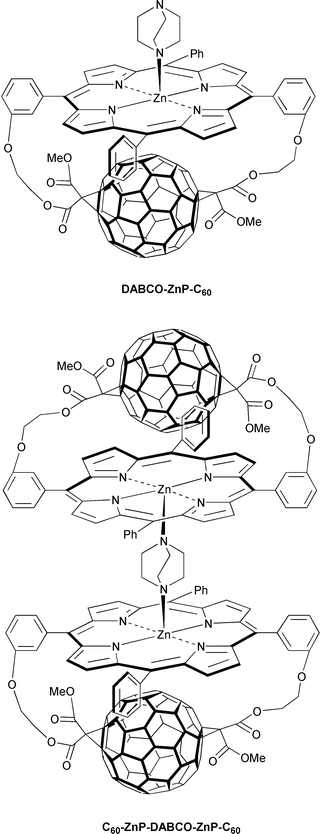
A different example involves a simplistic but powerful means to regulate donor–acceptor separations and orientations. More precisely, rigid, confined model ensembles are self-assembled, starting from a flexible ZnP-C60-ZnP system and DABCO.33 Similar to the simpler dyad ensembles (i.e., meta-ZnP-C60 and para-ZnP-C60) a photoinduced electron transfer evolving from the ZnP singlet excited state to the electron accepting fullerene governs the photophysics of the meta-ZnP-C60-ZnP and the more electron-rich para-ZnP-C60-ZnP. The resulting ZnP˙+-C60 ˙−-ZnP states decayed on a time scale of a few hundred nanoseconds to regenerate the ground state. For example, in o-dichlorobenzene the actual values are 150 ns and 290 ns in the meta-ZnP-C60-ZnP and para-ZnP-C60-ZnP isomers, respectively.
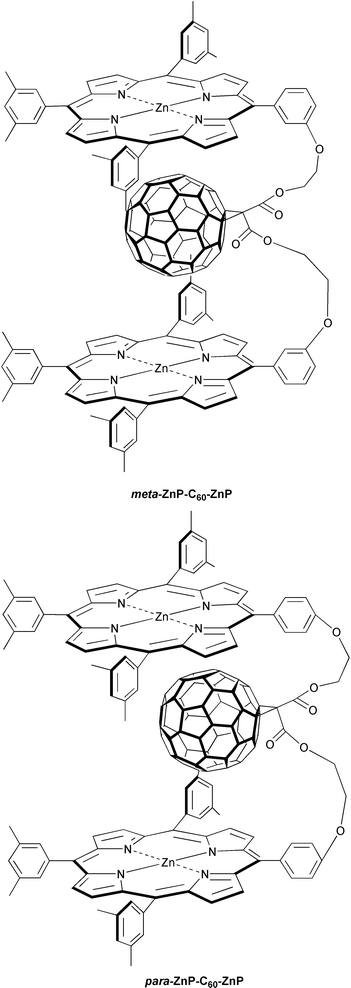
Addition of DABCO to toluene or o-dichlorobenzene solutions of meta-ZnP-C60-ZnP or para-ZnP-C60-ZnP evoked a strong reactivation of the 1*ZnP fluorescence (factor of ∼3). A similar impact was concluded from the transient absorption measurements, monitoring the decay and grow-in kinetics of 1*ZnP and ZnP˙+/C60˙− features, respectively. The sum of these effects evokes a model that infers the successful complexation of DABCO to the vacant sites of the two ZnP (i.e., dz2-orbitals) to expand the donor–acceptor separation considerably (i.e., triad versus tetrad). In polar media (i.e., o-dichlorobenzene) these readily available and stable ensembles are subject to a rapid intramolecular electron transfer to yield a long-lived radical pair with lifetimes close to a microsecond (∼700 ns). Again, the simple addition of a suitable component leads to a nearly five-fold improvement of the radical pair stability, besides an overall higher quantum yield of formation (Φ). Conversely, in a non-polar medium (i.e., toluene) energy transfer replaces electron transfer in deactivating the photoexcited chromophore, 1*ZnP, forming, in the final instance, the fullerene triplet excited state.

In retrospect, the reversible coordination of DABCO to ZnP-C60-ZnP creates a simple photoswitch, in which either an electron or energy transfer pathway deactivates the photoexcited ZnP. Larger DABCO concentrations, however, lead to the precipitation of poorly soluble oligomeric materials. It is interesting to note that the lifetime of the radical pair in these not well-defined structures gives rise to a further improvement (7.5 μs): a sufficiently fast separation of charges along the longitudinal axis is probably responsible for this consequence.
An appealing feature of the zinc-coordination is that the bond to an axially bond DABCO in a five-coordinated, square pyramidal Zn-complex is particularly labile. To prove that addition of a complexing agent will ultimately result in the destruction of the rigid donor–acceptor ensemble we added, for example, THF to an o-dichlorobenzene solution and probed the ZnP fluorescence. Indeed, a decrease in emission suggests the successful decomplexation, while removal of the volatile THF component led again to the reactivation of the emission. Unequivocally, this confirms the reversible transformation and, thereby, topological control over the donor–acceptor separation and orientation. Important for the performance of these fluorescing probes is the fact that, after probing this activation–deactivation cycle up to ten times, no notable deviation from the reversibility was found.
10 Future developments
Recent advances with respect to utilizing molecular recognition in the form of macrocyclic receptor molecules open new opportunities to control the dynamics of intramolecular events in the form of a supramolecularly assembled ZnP (i.e., porphyrin box, porphyrin cube and porphyrin tweezers)–C60 system.34 This constitutes certainly an elegant approach to assemble discrete van der Waals complexes bearing a close resemblance to natural photosynthesis.Interesting developments will also include the use of Th-hexadducts, in which the role of fullerene is to serve as the structure determining tecton.35 The highly symmetric Th-core facilitates loading of the fullerene core, for instance, with a shell of up to six electron donor/chromophore moieties, which renders these systems particularly appealing for photoconversion processes. Important features will include constructing larger light harvesting arrays and storing larger fractions of photonic energy in the charge-separated state.
Organic based photovoltaics, in which the knowledge gained during our more fundamentally oriented studies is used to advance the more technological issues, are expected to play an important role.36
11 Acknowledgements
This work was supported by the Office of Basic Energy Sciences of the U.S. Department of Energy. This is document NDRL-4338 from the Notre Dame Radiation Laboratory. I am deeply indebted to Professors Fukuzumi, Hirsch, Imahori, Maggini, Martin, Prato and Sakata for their productive collaborations and numerous stimulating discussions. This paper is dedicated to the memory of Ralf O. Guldi (1934-2001).12 References
- The Photosynthetic Reaction Center, ed. J. Deisenhofer and J. R. Norris, Academic Press, New York, 1993 Search PubMed.
- Electron Transfer in Chemistry Vol. I–V, ed. V. Balzani, Wiley-VCH, Weinheim, 2001 Search PubMed.
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 1999 Search PubMed.
- R. F. Curl, Angew. Chem., Int. Ed. Engl., 1997, 36, 1566 CrossRef.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593 CrossRef CAS.
- Fullerenes and Related Structures, ed. A. Hirsch, Top. Curr. Chem., Vol. 199, Springer, Berlin, 1999 Search PubMed.
- R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111 CrossRef.
- (a) S. Fukuzumi, I. Nakanishi, T. Suenobu and K. M. Kadish, J. Am. Chem. Soc., 1999, 121, 3468 CrossRef CAS; (b) S. Larsson, A. Klimkans, L. Rodriguez-Monge and G. Duskesas, J. Mol. Struct., 1998, 425, 155 CrossRef CAS.
- (a) D. M. Guldi, P. Neta and K.-D. Asmus, J. Phys. Chem., 1994, 98, 4617 CrossRef CAS; (b) D. M. Guldi and K.-D. Asmus, J. Am. Chem. Soc., 1997, 119, 5744 CrossRef CAS.
- Leading references: (a) M. M. Olmstead, D. A. Costa, K. Maitra, B. C. Noll, S. L. Phillips, P. M. Van Calcar and A. L. Balch, J. Am. Chem. Soc., 1999, 121, 7090 CrossRef CAS; (b) P. D. W. Boyd, M. C. Hodgson, C. E. F. Rickard, A. G. Oliver, L. Chaker, P. J. Brothers, R. D. Bolskar, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 1999, 121, 10487 CrossRef CAS.
- (a) M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Porphyrins Phthalocyanines, 2000, 4, 591 CrossRef; (b) J. Fujisawa, Y. Ohba and S. Yamauchi, Chem. Phys. Lett., 1998, 282, 181 CrossRef CAS; (c) D. M. Martino and H. van Willigen, J. Phys. Chem. A, 2000, 104, 10701 CrossRef CAS.
- P. A. Liddell, J. P. Sumida, A. N. MacPherson, L. Noss, G. R. Seely, K. N. Clark, A. L. Moore, T. A. Moore and D. Gust, Photochem. Photobiol., 1994, 60, 537 Search PubMed.
- Fullerene–porphyrin donor–acceptor dyads: (a) H. Imahori, K. Hagiwara, M. Aoki, T. Akiyama, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, J. Am. Chem. Soc., 1996, 118, 11771 CrossRef CAS; (b) T. D. M. Bell, T. A. Smith, K. P. Ghiggino, M. G. Ranasinghe, M. J. Shephard and M. N. Paddon-Row, Chem. Phys. Lett., 1997, 268, 223 CrossRef CAS; (c) P. S. Baran, R. R. Monaco, A. U. Khan, D. I. Schuster and S. R. Wilson, J. Am. Chem. Soc., 1997, 119, 8363 CrossRef CAS; (d) S. Higashida, H. Imahori, T. Kaneda and Y. Sakata, Chem. Lett., 1998, 605 CrossRef CAS; (e) K. Tamaki, H. Imahori, Y. Nishimura, I. Yamazaki, A. Shimomura, T. Okada and Y. Sakata, Chem. Lett., 1999, 227 CrossRef CAS; (f) R. Fong, D. I. Schuster and S. R. Wilson, Org. Lett., 1999, 1, 729 CrossRef CAS; (g) M. Wedel and F. P. Montforts, Tetrahedron Lett., 1999, 40, 7071 CrossRef CAS; (h) N. Armaroli, G. Marconi, L. Echegoyen, J.-P. Bourgeois and F. Diederich, Chem. Eur. J., 2000, 6, 1629 CrossRef CAS; (i) S. MacMahon, R. Fong II, P. S. Baran, I. Safonov, S. R. Wilson and D. I. Schuster, J. Org. Chem., 2001, 66, 5449 CrossRef CAS.
- Leading references for structural porphyrin analogs and references cited therein: (a) N. V. Tkachenko, L. Rantala, A. Y. Tauber, J. Helaja, P. H. Hynninen and H. Lemmetyinen, J. Am. Chem. Soc., 1999, 121, 9378 CrossRef CAS; (b) F. P. Montforts and O. Kutzki, Angew. Chem., Int. Ed., 2000, 39, 599 CrossRef CAS.
- For recent reviews on fullerene containing donor–acceptor ensembles and fullerene anions see (a) H. Imahori and Y. Sakata, Adv. Mater., 1997, 9, 537 CAS; (b) M. Prato, J. Mater. Chem., 1997, 7, 1097 RSC; (c) N. Martin, L. Sanchez, B. Illescas and I. Perez, Chem. Rev., 1998, 98, 2527 CrossRef CAS; (d) H. Imahori and Y. Sakata, Eur. J. Org. Chem., 1999, 2445 CrossRef CAS; (e) D. M. Guldi, Chem. Commun., 2000, 321 RSC; (f) D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS; (g) C. A. Reed and R. D. Bolskar, Chem. Rev., 2000, 100, 1075 CrossRef CAS; (h) D. Gust, T. A. Moore and A. L. Moore, J. Photochem. Photobiol. B, 2000, 58, 63 Search PubMed; (i) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS.
- Multiple fullerenes linked to a single porphyrin: (a) J.-F. Nierengarten, C. Schall and J.-F. Nicoud, Angew. Chem., Int. Ed., 1998, 37, 1934 CrossRef CAS; (b) J.-F. Nierengarten, L. Oswald and J.-F. Nicoud, Chem. Commun., 1998, 1545 RSC; (c) A. Rieder and B. Kräutler, J. Am. Chem. Soc., 2000, 122, 9050 CrossRef CAS.
- H. Imahori, K. Hagiwara, T. Akiyama, M. Aoki, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, Chem. Phys. Lett., 1996, 263, 545 CrossRef CAS.
- (a) E. Dietel, A. Hirsch, E. Eichhorn, A. Rieker, S. Hackbarth and B. Röder, Chem. Commun., 1998, 1981 RSC; (b) D. M. Guldi, C. Luo, M. Prato, E. Dietel and A. Hirsch, Chem. Commun., 2000, 373 RSC.
- (a) P. Cheng, S. R. Wilson and D. I. Schuster, Chem. Commun., 1999, 89 RSC; (b) D. I. Schuster, P. Cheng, S. R. Wilson, V. Prokhorenko, M. Katterle, A. R. Holzwarth, S. E. Braslavsky, G. Klihm, R. M. Williams and C. P. Luo, J. Am. Chem. Soc., 1999, 121, 11599 CrossRef CAS; (c) D. I. Schuster, Carbon, 2000, 38, 1607 CrossRef CAS.
- (a) T. Drovetskaya, C. A. Reed and P. D. W. Boyd, Tetrahedron Lett., 1995, 36, 7971 CrossRef CAS; (b) D. Kuciauskas, S. Lin, G. R. Seely, A. L. Moore, T. A. Moore, D. Gust, T. Drovetskaya, C. A. Reed and P. D. W. Boyd, J. Phys. Chem., 1996, 100, 15926 CrossRef CAS; (c) D. Gust, T. A. Moore and A. L. Moore, Res. Chem. Intermed., 1997, 23, 621 Search PubMed.
- (a) M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS; (b) Lecture Notes on Fullerenes Chemistry, ed. R. Taylor, Imperial College Press, London, 1999 Search PubMed.
- (a) C. Luo, D. M. Guldi, H. Imahori, K. Tamaki and Y. Sakata, J. Am. Chem. Soc., 2000, 122, 6535 CrossRef CAS; (b) H. Imahori, M. E. El-Khouly, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2001, 105, 325 CrossRef CAS; (c) H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607 CrossRef CAS; (d) H. Imahori, N. V. Tkachenko, V. Vehmanen, K. Tamaki, H. Lemmetyinen, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2001, 105, 1750 CrossRef CAS.
- Leading references and references cited therein: (a) A. Harriman, V. Heitz and J.-P. Sauvage, J. Phys. Chem., 1993, 97, 5940 CrossRef CAS; (b) H. Heitele, F. Pöllinger, T. Häberle, M. E. Michel-Beyerle and H. A. Staab, J. Phys. Chem., 1994, 98, 7402 CrossRef CAS; (c) A. Osuka, G. Noya, S. Taniguchi, T. Okada, Y. Nishimura, I. Yamazaki and N. Mataga, Chem. Eur. J., 2000, 6, 33 CrossRef CAS.
- (a) D. M. Guldi, C. Luo, M. Prato, A. Troisi, F. Zerbetto, M. Scheloske, E. Dietel, W. Bauer and A. Hirsch, J. Am. Chem. Soc., 2001, 123, 9166 CrossRef CAS; (b) D. M. Guldi, C. Luo, M. Prato, A. Troisi, F. Zerbetto, M. Scheloske, E. Dietel, W. Bauer and A. Hirsch, submitted.
- (a) K. Tamaki, H. Imahori, Y. Nishimura, I. Yamazaki and Y. Sakata, Chem. Commun., 1999, 625 RSC; (b) M. Fujitsuka, O. Ito, H. Imahori, K. Yamada, H. Yamada and Y. Sakata, Chem. Lett., 1999, 721 CrossRef CAS; (c) S. Fukuzumi, H. Imahori, M. E. El-Khouly, M. Fujitsuka, O. Ito and D. M. Guldi, J. Am. Chem. Soc., 2001, 123, 2571 CrossRef CAS.
- H. Imahori, D. M. Guldi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 6617 CrossRef CAS.
- (a) P. A. Liddell, D. Kuciauskas, J. P. Sumida, B. Nash, D. Nguyen, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1997, 119, 1400 CrossRef CAS; (b) D. Carbonera, M. Di Valentin, C. Corvaja, G. Agostini, G. Giacometti, P. A. Liddell, D. Kuciauskas, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1998, 120, 4398 CrossRef CAS; (c) J. L. Bahr, D. Kuciauskas, P. A. Liddell, A. L. Moore, T. A. Moore and D. Gust, Photochem. Photobiol., 2000, 72, 598 Search PubMed.
- D. Kuciauskas, P. A. Liddell, S. Lin, T. E. Johnson, S. J. Weghorn, J. S. Lindsey, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1999, 121, 8604 CrossRef CAS.
- T. Da Ros, M. Prato, D. M. Guldi, M. Ruzzi and L. Pasimeni, Chem. Eur. J., 2001, 7, 816 CrossRef CAS.
- (a) F. D’Souza, G. R. Deviprasad, M. S. Rahman and J.-P. Choi, Inorg. Chem., 1999, 38, 2157 CrossRef CAS; (b) F. D’Souza, G. R. Deviprasad, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 5277 CrossRef CAS.
- N. Armaroli, F. Diederich, L. Echegoyen, T. Habicher, L. Flamigni, G. Marconi and J.-F. Nierengarten, New J. Chem., 1999, 77 RSC.
- D. M. Guldi, C. Luo, T. Da Ros, M. Prato, E. Dietel and A. Hirsch, Chem. Commun., 2000, 375 RSC.
- D. M. Guldi, C. Luo, A. Swartz, M. Scheloske and A. Hirsch, Chem. Commun., 2001, 1066 RSC.
- (a) K. Tashiro, T. Aida, J.-Y. Zheng, K. Kinbara, K. Saigo, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 1999, 121, 9477 CrossRef CAS; (b) D. Sun, F. S. Tham, C. A. Reed, L. Chaker, M. Burgess and P. D. W. Boyd, J. Am. Chem. Soc., 2000, 122, 10704 CrossRef CAS.
- (a) E. Dietel, A. Hirsch, J. Zhou and A. Rieker, J. Chem. Soc., Perkin Trans. 2, 1998, 1357 RSC; (b) X. Camps, E. Dietel, A. Hirsch, S. Pyo, L. Echegoyen, S. Hackbarth and B. Röder, Chem. Eur. J., 1999, 5, 2362 CrossRef CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |