Laser control of chemical reactions
Niels E.
Henriksen
Department of Chemistry, Technical University of Denmark, DTU 207, DK-2800, Lyngby, Denmark
First published on 9th January 2002
Abstract
Chemical reactions are at the heart of chemistry and the dream of controlling the outcome of these reactions is an old one. Thus, with given reactants, a solvent and perhaps assisted by a catalyst, we would like to ‘steer’ the reactants into a particular desired product. This review focuses on how to control the dynamics of chemical reactions, beyond traditional temperature control, with the emphasis on unimolecular reactions. The electromagnetic radiation of lasers can induce so-called coherent dynamics. The recent theoretical and experimental results on this coherent control are explained and illustrated with computational and experimental examples.
 Niels E. Henriksen Niels E. Henriksen | Niels Engholm Henriksen was born in Vejle, Denmark in 1959. He received his PhD in chemical physics in 1987 from the Technical University of Denmark and a DSc in 1994 from the University of Copenhagen. After postdoctoral work at the University of Washington and the University of Copenhagen, he became assistant professor (1991) and associate professor (1995) at the Department of Chemistry, Technical University of Denmark. His research interests cover all theoretical aspects of molecular reaction dynamics, including time-dependent quantum mechanics, femtochemistry and laser control of chemical reactions, photofragmentation dynamics, gas–surface dynamics and more. |
1 Introduction
Chemistry is the study of the structure and properties of matter and of the reactions by which one form of matter may be produced from other forms. In this Review we will focus on some of the underlying physical principles which govern the dynamics of chemical reactions and, in particular, some recent theoretical and experimental developments concerning the control of chemical reactions via quantum mechanical interference effects.For a long time we have known that during an elementary chemical reaction, chemical bonds must break and form, typically, in a concerted way since activation energies (of bimolecular reactions) are smaller than bond dissociation energies. Furthermore, indirectly, say from traditional spectroscopy with infrared light, we have known that molecules can vibrate at very high frequencies ∼10−14 s = 10 fs. Thus, this is the typical time scale of nuclear motion and we can anticipate that the key events of chemical reactions will take place on a femtosecond time scale. However, only recently has it become possible to directly witness the motion of molecules as matter transforms from one form to another.1–6 The ultimate femtosecond resolution has been reached with the direct observation of chemical reactions as they proceed along the reaction path via transition states from reactants to products. [It should be remembered that the macroscopic rate of reaction which is measured for an ensemble of molecules at thermal equilibrium, typically, is much slower than the rate which can be inferred from the time it takes to cross the transition states because the fraction of reactants with sufficient energy to react is usually very small at typical temperatures.] This spectacular achievement was made possible by the development of femtosecond lasers, that is, laser pulses with a duration as short as a few femtoseconds.
The dynamics of elementary chemical reactions is followed in real time by femtosecond pump–probe spectroscopy.1–7 Thus, in a typical experiment two laser pulses are used, a ‘pump pulse’ and a ‘probe pulse’. The first femtosecond pulse initiates a chemical reaction, say the breaking of a chemical bond, and a second time-delayed femtosecond pulse probes this process. The ultrashort duration of the pump pulse implies that the zero of time is well-defined. The probe pulse is, for example, tuned to be in resonance with a particular transition in one of the fragments and when it is fired at a series of time-delays relative to the pump pulse, one can directly observe the formation of the fragment. This type of real-time chemistry is often called femtosecond chemistry (or simply, femtochemistry).
Another interesting aspect of femtosecond chemistry concerns the challenging objective of using femtosecond lasers to control the outcome of chemical reactions, say break a particular bond in a large molecule. As discussed in this Review, this type of control at the molecular level is much more selective than traditional methods of control where only macroscopic parameters like the temperature can be varied.
The dream of controlling the future of matter is an old one. In order to produce a desired product, chemists know how to choose proper starting materials, and how the yield of a particular product can be enhanced by a proper choice of solvents or by catalysts that selectively lower the activation barrier to the product. At this stage of the synthesis, the forces between molecules have been ‘selected’. In addition energy must be supplied, which is traditionally supplied as heat (or as photons in photochemistry). When we change the temperature, we change the average energy of all molecules in the sample. That is, we control the energy only on an average ‘macroscopic’ level. However, the way that energy is injected into the system affects the dynamics (i.e., the motion under the influence of the forces) and hence the outcome of the reaction.
We can supply energy into specific degrees of freedom, e.g., in the form of pre-excitation of the vibrational degrees of freedom of the reactants. The vibrational motion of polyatomic molecules encompasses all nuclei in the molecule and, as long as the displacement from the equilibrium configuration is sufficiently small, it can be broken down into the so-called normal mode vibrations. In special cases these vibrations take a particular simple form. Consider, e.g., a partially deuterated water molecule HOD. In this molecule, the H–OD and HO–D stretching motions are largely independent and the normal modes are, essentially, equivalent to the local bond stretching modes. To that end, consider the following reaction which has been studied theoretically as well as experimentally.
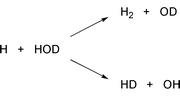 | (1) |
This is an illustration of the dynamics of ‘non-thermal’ states, that is, states which are inaccessible when energy is supplied to molecules in the form of heat. Clearly, the access to such states allows us to exert control beyond traditional means. A similar theme has been thoroughly studied in the so-called molecular beam experiments, where the initial state of the reactants can be fully specified. That is, the relative translational energy as well as the internal (vibrational/rotational) state of the reactants can be selected. The relative efficiency of translational and vibrational energy for overcoming energy barriers has, e.g., been studied. In some reactions, translational energy is very inefficient for promoting reaction compared to vibrational energy (see Ref. 9 and references therein).
It should be remembered that the composition of the reaction mixture at equilibrium is determined by the usual equilibrium constants K(T), unless, we are able to sustain the non-thermal distribution of the reactants.10
Ideally, we would like to be able to completely control the motion of each molecule in a given sample. Such complete ‘microscopic’ control of bimolecular reactions is very difficult, the orientation of the reactants can be controlled to some extent whereas it is virtually impossible to control the so-called impact parameter. Thus, in subsequent bimolecular collisions, we can have head on collisions, collisions where the molecules barely touch, and all variations in between. That is, it is difficult to establish completely identical initial configurations where the reactants have the same initial position. Much of the theoretical and experimental work on the control of chemical reactions has therefore focused on unimolecular reactions where the configuration of the initial state is well defined [this might also include long-lived intermediates of indirect bimolecular reactions]. To that end, consider the following reaction.
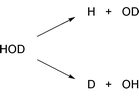 | (2) |
The two examples above illustrate how the dynamics of chemical reactions can be controlled by selective supply of energy to particular degrees of freedom prior to reaction. It is clear that a more general approach to control would involve an active intervention during the course of the reaction. That is femtosecond laser pulses are required.
Control of reactions at the microscopic level is one of the hot research areas of chemical physics. The aim is to control the outcome of a reaction by means of external laser fields such that the yield of a particular product is enhanced.13–19 That is, the problem is, given some initial state, how to guide the system into a desired final state.
This Review is organized in the following way. In Section 2, a comparison of the basic concepts of laser control and traditional temperature control is given. This discussion includes an elementary explanation and definition of concepts such as the Boltzmann distribution and incoherent superpositions of stationary states versus coherent superpositions of stationary states and quantum interference. In Section 3, some applications of laser control to chemical reactions are presented. Finally, Section 4 gives a short summary and outlook.
2 Incoherent versus coherent excitation
According to quantum mechanics, the vibrational and rotational energy states of molecules are quantized, which implies that only discrete molecular energy levels can be found. Traditionally, energy is supplied as heat and the molecular energy states are populated in a so-called incoherent way according to the Boltzmann distribution. The energy states can be populated in other ways. So-called coherent excitation of the molecular energy states is the basis of (coherent) laser control where one takes advantage of quantum interference effects. These concepts are, briefly, discussed in this Section.2.1 Incoherent excitation
According to quantum mechanics, the allowed energy eigenstates of an isolated molecule are described by wave functions, Ψn(x,t) = ψn(x)e−iEnt/ℏ, where ℏ ≡ h/(2π), h is Planck’s constant, and En is the energy of the state. The wave functions depend on the time, t, and coordinates, x, which specify the configuration of the molecule. The molecule can be found in any of these energy states, where n = 0, 1, 2, ... At T = 0 K, all molecules will be found in the lowest energy state with the energy E0. Higher temperatures correspond to molecular motion and via inelastic collisions, that is processes such as| A + BC(n) → A + BC(m) | (3) |
| pn = Q−1e−En/(kBT) | (4) |
When molecules are produced in chemical reactions, the energy states are not populated according to the Boltzmann distribution. However, this distribution will quickly be established through collisional energy transfer. Thus, say at 1 atm, the Boltzmann distribution will typically be established among the vibrational energy levels within the order of a millisecond and within the order of a nanosecond among the rotational energy levels. When the pressure is reduced, the Boltzmann distribution will, however, be established more slowly. In liquids, the Boltzmann distribution will typically be established among the vibrational energy levels within the order of a picosecond.
Consider, as an example, the probability of finding the system at the configuration specified by the coordinate x, given by
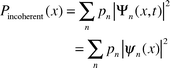 | (5) |
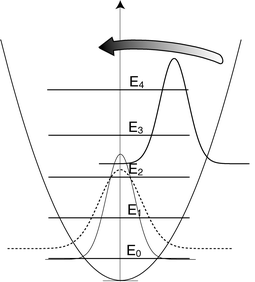 | ||
| Fig. 1 Incoherent versus coherent excitation of a harmonic oscillator. The solid (thin) line is the probability distribution associated with the ground state wave function, |ψ0(x)|2. The dashed line is the Gaussian probability distribution associated with thermal excitation at temperature T. The displaced Gaussian corresponds to a coherent excitation of the quantum states of the harmonic oscillator. The arrow indicates that this wave packet is vibrating at the frequency of the oscillator. The position of the baselines (where the value of the Gaussians approach zero) reflects their average energy, which for the ground state is the zero-point energy E0. | ||
When we consider a thermally activated unimolecular reaction, say the fragmentation of a triatomic molecule with two product channels,
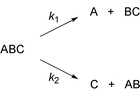 | (6) |
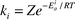 or transition state (RRKM) theory
or transition state (RRKM) theory 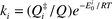 where Q and Q‡i are partition functions associated with the reactant and the activated complex leading to product channel i, respectively, and Ei0 are the barrier heights associated with the product channels. Thus, when we neglect the weak temperature dependence of the pre-exponential factors, b = k1/k2
∼
e−ΔEa/RT , where ΔEa = E1a
−
E2a is the difference between the activation energies (∼ barrier heights) of the two channels.
where Q and Q‡i are partition functions associated with the reactant and the activated complex leading to product channel i, respectively, and Ei0 are the barrier heights associated with the product channels. Thus, when we neglect the weak temperature dependence of the pre-exponential factors, b = k1/k2
∼
e−ΔEa/RT , where ΔEa = E1a
−
E2a is the difference between the activation energies (∼ barrier heights) of the two channels.
This ratio can be controlled to some extent, by the temperature, provided ΔEa ≠ 0. Thus, when T is small, that is ΔEa ≫ RT, then b ≪ 1 when ΔEa > 0 and b ≫ 1 when ΔEa < 0. On the other hand, at higher temperatures both channels will be populated more evenly. Thus in the limit where T is large, that is ΔEa ≪ RT, then b ∼ 1.
2.2 Coherent excitation
It is possible to create a coherent linear superposition of energy eigenstates, e.g., via laser excitation. This is a state of the form ΣncnΨn(x,t), with a particular phase relation between the coefficients cn. The value of these coefficients is determined by the pulsed laser excitation.We consider again the probability of finding the system at the configuration specified by the coordinate x, then according to the rules of quantum mechanics,
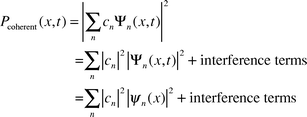 | (7) |
| Pcoherent (x,t) = |c1|2|ψ1(x)|2 + |c2|2|ψ2(x)|2+ 2|c1||c2|| ψ1(x)||ψ2 (x)|cos[(ΔEt/ℏ) − δ] | (8) |
Note that the interference term becomes time-independent when the two states are degenerate, that is ΔE = 0. This observation is used in one of the two major schemes for coherent control.18,19 In the other scheme,14,15 which we will focus on in this Review, the time evolution of quantum mechanical states plays an important role.
The time evolution in eqn. (7) is described by the time-dependent Schrödinger equation, provided the molecule is isolated from the rest of the universe. In practice, there are always perturbations from the environment, say due to inelastic collisions as in eqn. (3). The coherent sum in eqn. (7) will then relax to the incoherent sum of eqn. (5), that is, the off-diagonal interference terms will vanish and |cn|2 → pn corresponding to the Boltzmann distribution. As mentioned earlier, the relaxation time depends on the pressure. In order to take advantage of coherent dynamics it is, of course, crucial that relaxation is avoided within the duration of the relevant chemical reaction.
3 Applications
3.1 Orientation of molecules
In bimolecular reactions, the orientation of reactants can play an important role (see Ref. 9 and references therein). For example, in the reaction of alkali atoms M with CH3I to give MI + CH3, it was found that reaction preferentially occurred when M collided with the I end of the CH3I molecule.As an illustration of the difference between incoherent (thermal) and coherent (laser) excitation, consider the orientation of a linear molecule. The rotational energy eigenstates are in this case given by YJM(θ,ϕ), the so-called spherical harmonics where the two angles, θ and ϕ, specify the orientation of the molecule and J and M are the two quantum numbers which label the states (recall, e.g., the angular dependence of the orbitals of the hydrogen atom). Eqn. (5) takes in this case the form,
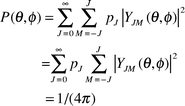 | (9) |
On the other hand,20 a coherent superposition of the two states YJM(θ,ϕ ) with J = 0, 1 and M = 0 is illustrated in Fig. 2. Here a preferred orientation is obtained (note that the orientation is time-dependent, according to eqn. (8)).
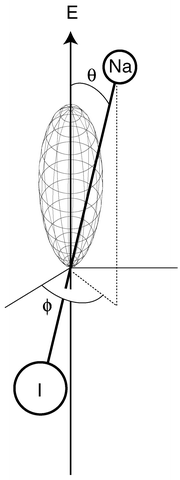 | ||
| Fig. 2 An oriented NaI molecule. The figure shows the angular part of the nuclear wave function which depends on the two angles θ and ϕ. The wave function is shown in a so-called polar plot where the distance from the origin to the surface is equal to the magnitude of the orientation dependent wave function. Note that there is a high probability of finding the Na atom pointing upwards along the vertical axis, which is the polarization direction of the linearly polarized electric field (E) of the laser pulse which created this state. | ||
3.2 Selective bond breaking
Consider, as an example, again the unimolecular decomposition of HOD in eqn. (2). First, we note that the channel H + OD is favoured in thermal decomposition, and in photodissociation out of the vibrational ground state via the first electronically excited state, the H + OD channel also dominates at all photon energies.11As mentioned in the Introduction, photochemical decomposition of vibrationally pre-excited HOD gives a large degree of control. The coherent excitation of vibrational wave packets gives, however, the ultimate control in this and similar reactions.14,21 One way to control the outcome of the reaction is to use two laser pulses, where the first pulse forces the molecule into a non-stationary vibrational state and the second pulse initiates the reaction at an appropriate time delay relative to the first pulse. An ultrashort pulse in the ultraviolet region of the spectrum can launch a localized wave packet on a potential energy surface at a well-defined time. The principle is illustrated in Fig. 3. This scheme has a close formal correspondence to femtosecond pump–probe spectroscopy. It should be noted that the HOD example would be very difficult to study experimentally since the vibrational dynamics is very fast, with a vibrational period about 10 fs. Thus, laser pulses with durations below 10 fs are required, and such pulses are at present not readily available.
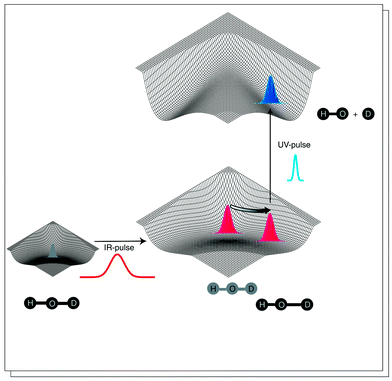 | ||
| Fig. 3 Selective bond breakage in the HOD molecule. The potential energy surfaces in the figure correspond to the electronic ground state and the first electronically excited state which supports no bound states. Initially, the molecule is in the vibrational ground state, described by the wave function with the Gaussian shape (lower left corner). An intense pulse in the infrared region can induce vibrational motion in the molecule, here HO–D stretching (this excitation method works only, without complications, when it is used on pre-oriented molecules). An ultrashort laser pulse in the ultraviolet region of the spectrum is fired when the wave packet is located such that the dissociation dynamics evolve solely in the desired product channel. | ||
This scheme can readily be extended beyond the case where the normal mode vibrations coincide with local bond stretching modes.22 We note in particular that the short duration of the second (control) pulse implies that intramolecular vibrational relaxation (IVR) is bypassed. The scheme considered here is, however, based on a limited number of optimization parameters, primarily the center frequencies and durations of the laser pulses, and the time delay between the two pulses. In more complex systems these parameters may not be sufficient and, furthermore, the separation of the laser pulse into two distinct non-overlapping pulses might not be possible.
3.3 Selective preparation of chiral molecules
The problem of laser controlled chemical reactions is in general how to design a laser pulse which can guide the system into the desired final state. In the previous subsection, we considered a problem where a suitable pulse sequence could be ‘designed’ by an educated guess. In general, it is not possible to guess what form an optimal pulse takes. The problem can, however, be handled systematically by an optimization technique called Optimal Control Theory.23 This leads to a procedure where the time-dependent Schrödinger equation must be solved iteratively.This technique has, for example, been applied to the selective preparation of chiral molecules (enantiomers) from a racemic mixture, i.e., an equal mixture of R- and L-enantiomers. That is, the problem of how to transform a racemic mixture into an optically pure form (see Ref. 24 and references therein).
An application to a racemic mixture of pre-oriented H2POSH at low temperatures, has been considered.24 The two enantiomers are shown in Fig. 4. They corresponds to two different conformations associated with the torsional motion of SH around the PS bond. The potential energy associated with this motion takes the form of a double-well potential with equivalent minima corresponding to the two enantiomers and a maximum when the SH and PO bonds are eclipsed. A pure enantiomer exists when all of the population is localized in one side of the double-well potential. It should be noted that the lifetime of such an enantiomer is very short compared to more conventional enantiomers where chemical bonds must be broken in order to transform the molecule into its mirror image. Thus, in the present case the lifetime is just 630 ps, which is equal to the tunneling time between the two localized states.
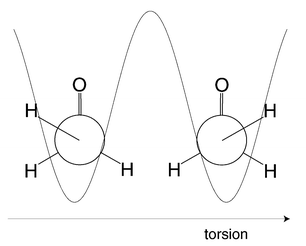 | ||
| Fig. 4 Enantiomers of H2POSH, viewed along the SP bond, and the double-well potential corresponding to the torsional motion. Using a specially designed sequence of circularly polarized laser pulses, an equal (racemic) mixture of the enantiomers could be transformed into an optically pure form, where only one of the enantiomers is present.24 | ||
The optimized electric field corresponds to a circularly polarized pulse with a duration of ∼100 ps and a central frequency of ∼200 cm−1. This pulse was designed without any assumptions concerning the analytical form of the electric field, thus the optimization goes beyond the variation of a few parameters in the laser pulse.
3.4 Laboratory feedback control
As pointed out above, in order to reach a specific reaction channel, the electric field of the laser pulse must be specifically designed to the molecular system. All features of the molecular system must, however, be completely known in order to solve this problem. The full Schrödinger equation for a large molecular system with many electrons and nuclei can at present only be solved in an approximate way [the present limits are at a reaction like the one in eqn. (1), see Ref. 25]. Thus, in practice, the precise form of the laser field cannot always be calculated in advance.In order to bypass this problem a clever idea has been introduced: the laboratory feedback control technique.26,27 The optimization procedure is based on the feedback from the observed experimental signal (e.g., a branching ratio) in an algorithm that iteratively improves the applied femtosecond laser pulse. This iterative optimum-seeking process, has been termed ‘training lasers to be chemists’.28 This captures the spirit of the method, however, it must be remembered that a laser can do one thing which chemists and their Bunsen burner cannot do; it can create coherent dynamics. This automated experimental implementation of coherent laser control has been applied to the reaction,
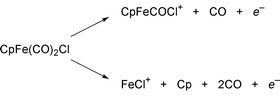 | (10) |
4 Summary and outlook
We have discussed coherent (laser) control for simple unimolecular (photodissociation) reactions, and emphasized how this approach differs from traditional temperature control of the reaction dynamics. In chemistry there is almost always some interplay between theory and experiment. To that end it is interesting to note that this field started with purely theoretical predictions which were subsequently verified experimentally.We have focused on highly relevant objectives such as selective bond breaking and selective preparation of chiral molecules. However other objectives can also be envisioned like: selective fragment orientation, i.e., the control of the spatial distribution of fragments;29 and selective electron transfer, i.e., the control of which nuclei the valence electrons will follow when a specific chemical bond is broken.30 With the demonstration of the laboratory feed-back control technique many interesting applications can be anticipated. For example, the investigation of coherent control in organic photochemistry31 and the control of chemical reactions in condensed (liquid) phases.
Will these new techniques ever move out of the research laboratories? It might turn out to be a valuable tool for some highly specialized applications, say like photodynamic cancer therapy, but at present the costs are probably in most cases much too high compared to the benefits. However, it is not easy to predict future developments, and laser control might one day turn out to be a valuable tool also in the chemical and pharmaceutical industry. In the mean time, studies on laser control give us additional insight into the inner workings of chemical reactions.
5 Acknowledgement
This work was supported by The Danish Natural Science Research Council.6 References
- A. H. Zewail, Femtochemistry, World Scientific, Singapore, 1994, vols. 1, 2 Search PubMed.
- Femtosecond Chemistry, ed. J. Manz and L. Wöste, VCH, Weinheim, 1995 Search PubMed.
- Femtochemistry, ed. M. Chergui, World Scientific, Singapore, 1996 Search PubMed.
- Femtochemistry and Femtobiology, ed. V. Sundström, Imperial College Press, London, 1997 Search PubMed.
- Chemical Reactions and their Control on the Femtosecond Time Scale, XXth Solvay Conference on Chemistry, ed. P. Gaspard and I. Burghardt, Adv. Chem. Phys., 1997, 101 Search PubMed.
- A. H. Zewail, J. Phys. Chem., 2000, 104, 5660 CrossRef CAS.
- N. E. Henriksen and V. Engel, Int. Rev. Phys. Chem., 2001, 20, 93 CrossRef CAS.
- A. Sinha, M. C. Hsiao and F. F. Crim, J. Chem. Phys., 1990, 92, 6333 CrossRef CAS.
- R. N. Zare, Science, 1998, 279, 1875 CrossRef CAS.
- N. E. Henriksen, G. D. Billing and F. Y. Hansen, J. Phys. Chem., 1992, 96, 223 CrossRef CAS.
- D. G. Imre and J. Zhang, Chem. Phys., 1989, 139, 89 CrossRef CAS.
- N. E. Henriksen, Adv. Chem. Phys., 1995, 91, 433 Search PubMed.
- A. H. Zewail, Physics Today, 1980, 27 Search PubMed.
- D. J. Tannor and S. A. Rice, J. Chem. Phys., 1985, 83, 5013 CrossRef CAS.
- S. A. Rice and M. Zhao, Optical control of Molecular Dynamics, Wiley, 2000 Search PubMed.
- B. Kohler, J. L. Krause, K. R. Wilson, V. V. Yakovlev, R. M. Whitnell and Y. Yan, Acc. Chem. Res., 1995, 28, 133 CrossRef CAS.
- J. Manz, in Femtochemistry and Femtobiology: Ultrafast Reaction Dynamics at Atomic-Scale Resolution, ed. V. Sundström, Imperial College Press, London, 1997 Search PubMed.
- P. Brumer and M. Shapiro, Chem. Phys. Lett., 1986, 126, 541 CrossRef CAS.
- M. Shapiro and P. Brumer, J. Chem. Soc., Faraday Trans., 1997, 93, 1263 RSC.
- N. E. Henriksen, Chem. Phys. Lett., 1999, 312, 196 CrossRef CAS.
- B. Amstrup and N. E. Henriksen, J. Chem. Phys., 1992, 97, 8285 CrossRef CAS.
- B. Amstrup and N. E. Henriksen, J. Chem. Phys., 1996, 105, 9115 CrossRef CAS.
- A. P. Peirce, M. A. Dahleh and H. Rabitz, Phys. Rev. A, 1988, 37, 4950 CrossRef.
- K. Hoki, Y. Ohtsuki and Y. Fujimura, J. Chem. Phys., 2001, 114, 1575 CrossRef CAS.
- D. H. Zhang, M. Yang and Soo-Y. Lee, J. Chem. Phys., 2001, 114, 8733 CrossRef CAS.
- R. S. Judson and H. Rabitz, Phys. Rev. Lett., 1992, 68, 1500 CrossRef CAS.
- A. Assion, T. Baumert, M. Bergt, T. Brixner, B. Keifer, V. Seyfried, M. Strehle and G. Gerber, Science, 1998, 282, 919 CrossRef CAS.
- R. F. Service, Science, 1998, 279, 1847 CrossRef CAS.
- M. Machholm and N. E. Henriksen, J. Chem. Phys., 1999, 111, 3051 CrossRef CAS.
- M. Grønager and N. E. Henriksen, J. Phys. Chem., 1998, 102, 4277 CrossRef CAS.
- R. J. Levis, G. M. Menkir and H. Rabitz, Science, 2001, 292, 709 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |