DOI:
10.1039/B103904K
(Review Article)
Chem. Soc. Rev., 2002,
31, 12-21
Nucleophilic transition metal based cyclization of allenes
Received
7th September 2001
First published on 11th December 2001
Abstract
Allenes bearing a pro-nucleophile can be cyclized on treatment with a wide variety of transition metal catalysts and reagents: palladium, cobalt, ruthenium, silver, rhodium, lanthanides, gold. The nucleophilic groups can be nitrogen, oxygen or carbon based and can form rings of various sizes, often with good control of stereochemistry. A variety of mechanisms can be proposed for these reactions and the metal complex can be used to introduce a variety of functional groups during cyclization. Several heterocyclic natural products have been prepared using a selection of these reactions.
| Roderick Bates was born in 1965 in Hampshire, England. He received a BSc from Imperial College, London, and developed a taste for transition metal chemistry. He stayed there to obtain a PhD under the guidance of Professor S. V. Ley. Following a spell as an SERC-NATO Postdoctoral fellow with Professor L. S. Hegedus at Colorado State University, he joined the faculty of the University of North Texas and then of Chulalongkorn University, Bangkok, Thailand. He is now a researcher at the Chulabhorn Research Institute in that city. |
 Vachiraporn Satcharoen Vachiraporn Satcharoen | Miss Vachiraporn Satcharoen was born in Bangkok in 1976. At Chulalongkorn University, she received her BS in 1998 and her MS in 2000 on the subject of diastereoselective cyclization of allenes. She is now working as an Instructor in the Department of Chemistry of Naresuan University in Phitsanulok, Thailand. |
1 Introduction
The attack of a nucleophilic species onto an unsaturated moiety has its origin in the Wacker process. Since then the equivalent intramolecular reactions have also been investigated. For the most part, these investigations have focused on alkenes, 1,3- dienes and alkynes as the unsaturated reaction partners.1 Allenes have also been examined and show a number of advantages. Allenes can be synthesized by a number of procedures.2
Generally, in such reactions, one of the allene double bonds remains in the product and is useful for further chemistry, especially with the growth of alkene metathesis reactions. In a number of cases, allenes have been found to be more reactive than other unsaturated groups. 1,3-Disubstituted allenes are also inherently chiral and can be prepared with high enantiomeric excess by a number of methods, although only a few of the reactions discussed here take advantage of this. This review is limited to the cyclization of an allene by intramolecular attack of a nucleophile (using the term loosely) induced by a d- or f-block metal ion or complex.3
An additional interesting facet of these allene reactions is the mechanism. Three principal mechanisms have been put forward and in most cases there is little hard evidence available. In the presence of an electrophilic metal complex, such as palladium(II), an η2-complex might form with one of the allene double bonds (Scheme 1). This would be expected to be electrophilic and could be attacked by a pendant nucleophile at either coordinated carbon. This is directly analogous to the Wacker reaction. Thus four possible η1-complexes could be formed, of which two are in equilibrium with the same η3-complex. It is also possible that the η2-complex would react with ligands (such as halides) on the metal, leading to other intermediates and pathways.
 |
| | Scheme 1 | |
The organic products of the reaction would then come from a subsequent reaction of the η1- or η3-complex, such as reductive elimination (if the metal has a σ-bonded ligand), protonolysis or CO insertion.
A second alternative is possible when the metal complex employed has a σ-bonded ligand (for example, H, R or halogen) (Scheme 2). Insertion of the allene might then generate an η3-allyl complex. This could cyclize by nucleophilic attack on either terminus. In many cases either the η2- or the η3- mechanism can be drawn leading to the same product.
 |
| | Scheme 2 | |
In cases where R is a nucleophilic atom (such as a halide ion), related intermediates can arise by intramolecular nucleophilic attack of R− onto the coordinated allene, leading to the same η3-complex. This pathway presumes that the resident nucleophile, X, is weakly reactive.
The third alternative is that the metal complex initially interacts with the ‘nucleophilic’ group; this is followed by insertion and reductive elimination (Scheme 3). Insertion may involve either of the allene double bonds to generate different ring sizes.
 |
| | Scheme 3 | |
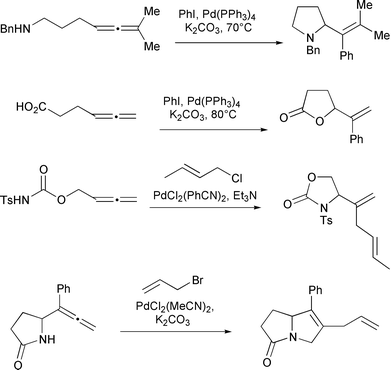 |
| | Scheme 4 | |
Mechanisms involving both attack on an η2- and an η3-palladium complex can be drawn (see Schemes 1 and 2). There is a consensus that the first step is oxidative addition of palladium(0) to the organic halide to generate an organopalladium(II) species that acts as the cyclization trigger. Beyond that, there is little evidence to indicate whether an η2-mechanism (as in Scheme 1) or an η3-mechanism (as in Scheme 2) operates. Indeed, there may not be a single general mechanism for all of these reactions.
An unusual result has been reported by Hiemstra involving attack on the central allene carbon (Scheme 5).9 In this process, at least, an η2-complex is likely to be the key intermediate, as attack on the central carbon of an η3-allyl complex is infrequently observed, especially with soft phosphine ligands.
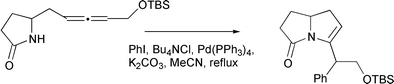 |
| | Scheme 5 | |
A number of ring sizes can be made. Both Kang and Hiemstra have used allene (1) (Scheme 6).10,11 Attack of the nucleophile on the near carbon of the allene would lead to an azetidine; attack on the far carbon would lead to a tetrahydropyridine. Hiemstra has proposed that the mechanistic pathways diverge according to the geometry of an η3-allyl intermediate. In the syn conformation (2), only the azetidine (3), the kinetic product, can form. In the anti conformation (4) (aminoalkyl side chain is endo-), a route to the tetrahydropyridine (5), the thermodynamic product, is open. The azetidine (3), however, can reenter the process due to its ring strain, and, with an extended reaction time or a higher temperature, be converted to the tetrahydropyridine.
 |
| | Scheme 6 | |
The reaction has also been reported to lead to vinyl epoxides12 and to vinyl aziridines.13 The five membered rings (by endo-cyclization) are the alternative products, and the outcome appears to be highly dependent on the reaction conditions (Scheme 7).
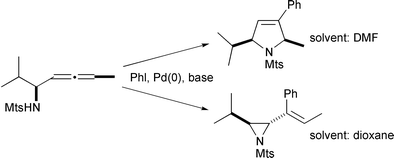 |
| | Scheme 7 | |
3 Palladium catalyzed cyclocarbonylation
Similar reaction mixtures to those outlined above, but in the presence of carbon monoxide, result in the corresponding ketones (Scheme 8). Walkup has been able to form tetrahydrofurans from γ-hydroxyallenes in this way,14 while Kang has converted α-sulfonamido allenes to dihydropyrrolidines in a carbonylative endo-cyclization. Similarly γ- and δ-sulfonamidoallenes were converted to pyrrolidines and piperidines respectively.15 Kang has put forward a mechanism involving an η3-allyl intermediate for this transformation.
 |
| | Scheme 8 | |
This reaction may be considered a variation on those discussed in Section 2: CO insertion follows oxidative addition, resulting, in the end, with a ketone.
4 The oxidative PdX2 system
For some years, Bäckvall's group has been studying the reaction of 1,3-dienes with palladium(II)/halide ion systems. The same protocol is successful with allenes (i.e. 1,2-dienes) (Scheme 9).16 The products are the cyclized vinyl bromides; alcohols, carboxylic acids and several nitrogen derivatives could be employed as nucleophiles, although a simple amine gave a poor yield. In addition, N-tosyl amides were converted to oxazolidinones in good yields.
 |
| | Scheme 9 | |
Mechanistic studies indicate the involvement of both η2- and η3- intermediates. Thus, an η2-allene palladium complex is attacked at the centre carbon by halide as an external nucleophile. This leads to an η3-allyl complex which undergoes cyclization. Indeed, the η3-complex (6) could be prepared in good yield and was shown to cyclize on exposure to base at a reasonable rate (Scheme 10).
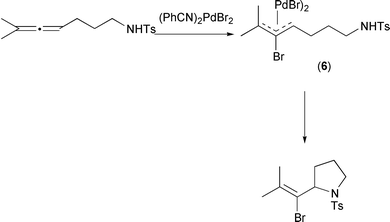 |
| | Scheme 10 | |
5 Pd(II)–CO–alcohol system leading to substituted acrylates
Use of palladium(II), an alcohol (typically methanol) and carbon monoxide leads to substituted acrylates as the cyclization products. A mechanism can be proposed involving η2-coordination of the allene to palladium, followed by nucleophilic attack of the nucleophilic atom leading to an η1-vinyl palladium species. CO insertion, followed by alcohol attack yields the product. As this involves a net reduction of palladium, catalysis is maintained by addition of Cu(II) as an oxidising agent.17 (Scheme 11)
 |
| | Scheme 11 | |
The corresponding reaction with oxygen nucleophiles has also been reported: Walkup has shown that allenic aldehydes, perhaps as their hemiacetals formed in situ, cyclize to give furanosides with excellent diastereoselectivity (Scheme 12).18 The cyclization is also successful with alcohol nucleophiles. In the case of tetrahydrofuran formation, however, better stereoselectivity was obtained by a mercury induced cyclization of silyl ethers, followed by carbonylation of the organomercurial (Scheme 13).19
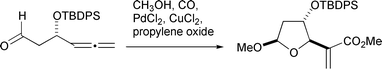 |
| | Scheme 12 | |
 |
| | Scheme 13 | |
In contrast, Snider and He found that six-membered ring formation using the palladium methodology was highy satisfactory (Scheme 14).20 Thus, cyclization of the allene (7), prepared in four steps from farnesyl bromide, yielded a 6 ∶ 1 mixture of the equatorial and axial isomers (8). The desired isomer, after purification, could be hydrolysed to rhopaloic acid, a cytotoxic marine sponge natural product.
 |
| | Scheme 14 | |
6 Acyl cobalt methodology
Acyl cobalt complexes can be generated by the reaction of NaCo(CO)4 with reactive alkyl halides, especially methyl iodide. This has placed some limitation on the available substrates, although other acyl cobalts are available from reactions with acid chlorides or with ketenes. This is in contrast to the palladium chemistry involved in Section 3 which works best with aryl substrates. Addition of the acyl complexes to a variety of allenes bearing pro-nucleophilic groups in the γ-position results in formation of an η3-allyl complex (9) which then cyclizes to a five membered ring (Scheme 15).21 This has been demonstrated for O, N and C nucleophiles. The last of these groups is rarely studied in allene–metal chemistry.
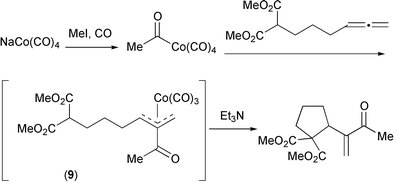 |
| | Scheme 15 | |
The reaction also works for both possible kinds of disubstituted allenes (Scheme 16). 1,3-Disubstituted allenes are stereospecifically converted to the E-products. 1,1-Disubstituted allenes cyclize efficiently to generate a quaternary centre. Trisubstituted allenes failed to react.
 |
| | Scheme 16 | |
 |
| | Scheme 17 | |
8 Cycloisomerization
Silver(I) has been known for some time to be an electrophilic trigger for allene cyclization with both O- and N-nucleophiles.23 The reaction may be extended to N-tosyl carbamates if a base, such as triethylamine, is included (Scheme 18).24
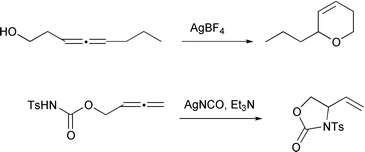 |
| | Scheme 18 | |
Oximes are also competent nucleophiles (Scheme 19). Their cyclization generates a nitrone. In some cases, these may be sufficiently stable to be isolated. They may also be trapped in situ by alkenes.25
 |
| | Scheme 19 | |
The high and predictable stereoselectivity of the nitrone cycloaddition makes this a synthetically useful process. The unnamed ant-venom alkaloid (10) was prepared using this method (Scheme 20). Treatment of the allene (11) with silver(I) and then MVK gave the cycloadduct (12) as a mixture of epimers due to the chiral centre α to the ketone. Catalytic hydrogenation then reduced the double bond, cleaved the O–N bond and reduced the iminium ion formed from the newly released amine and ketone groups. Excision of the alcohol group then yielded the pyrrolizidine target.
 |
| | Scheme 20 | |
Marks and McDonald have shown that lanthanide derivatives can catalyse the cycloisomerization of γ-and δ-aminoallenes (Scheme 21).26 In the case of monosubstituted γ-allenes, mixtures of tetrahydropyridines and pyrrolidines were formed, resulting from insertion of either allene double bond into a nitrogen–lanthanide bond (Scheme 22). The homologous δ-aminoallenes yielded only the piperidines. More highly substituted γ-aminoallenes were processed more selectively, leading to only the pyrrolidine products. An alkyl group adjacent to the nitrogen atom resulted in diastereoselective cyclization, resulting in trans-pyrrolidines and cis-piperidines.
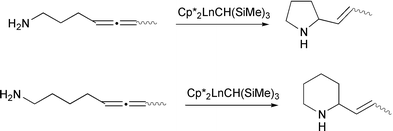 |
| | Scheme 21 | |
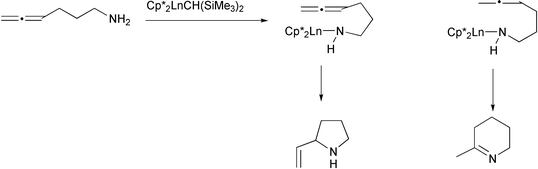 |
| | Scheme 22 | |
While a series of lanthanides were found to be effective in the cyclization reaction, the metal with the highest turnover frequency was found to be yttrium (although it is not strictly a lanthanide) whose ionic radius is part way between the largest, lanthanum, and the smallest, lutetium. This is an excellent example of the fine-tuning possible with the f-block elements.
Pyrrolidine 197B (13) has been prepared using this aminoallene cylization (Scheme 23).
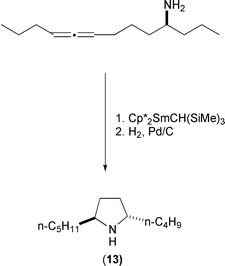 |
| | Scheme 23 | |
The synthesis of a bicyclic alkaloid, of which many occur in nature, might then be achievable by an extension of this: the use of an amine with two unsaturated substituents. The synthesis of xenovenine (14) is an example of this, using a tandem cyclization (Scheme 24). On exposure of allene (15) to a range of bis-Cp* lanthanide species, however, cyclization only onto the allene was observed. This is consistent with the observation that allenes are some twenty times more reactive than alkenes and, perhaps, that secondary amines are more hindered than primary. The solution to this problem was arrived at by employing ‘constrained geometry’ pre-catalysts. These species, such as samarium complex (16), in which one Cp* ring has been replaced by a chelating amino-arm, offer a less crowded reaction site and higher reactivity.27 This resulted in successful bis-cyclization to (17).
The natural product (14) was then obtained by reduction of the remaining double bond.
 |
| | Scheme 24 | |
Yamamoto has described two palladium catalyzed cyclo-isomerizations that proceed via allene insertion (Scheme 25). Treatment of allenes bearing a variety of nitrogen pro-nucleophiles with a palladium catalyst resulted in cyclization onto the nearer of the two double bonds, to give five or six membered rings. The presence of acetic acid was found to be useful for good reaction rates and yields.28
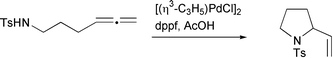 |
| | Scheme 25 | |
Similarly, treatment of allenic malonates with a palladium catalyst yields the corresponding cyclic products.29 This reaction is similar to one reported by Trost et al. for the synthesis of macrocycles by C–C bond formation (Scheme 26).30
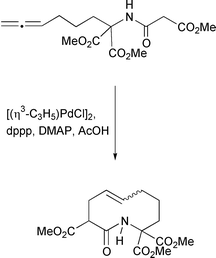 |
| | Scheme 26 | |
A variation on this theme involves the use of carbon monoxide. With ruthenium catalysts it has proved possible to intercept the pro-nucleophile with CO prior to cyclization (Scheme 27). This has resulted in the formation of a series of γ- and δ-lactones and lactams.31,32
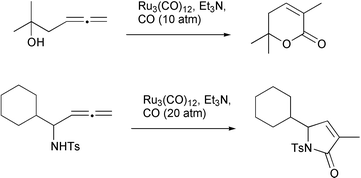 |
| | Scheme 27 | |
In the case of the sulfonamides, a mechanism was proposed involving oxidative addition of ruthenium to the N–H bond, followed by insertion of an allenic double bond into the Ru–H bond to form a vinyl organometallic (Scheme 28). Carbon monoxide insertion and reductive elimination then completed the sequence.
 |
| | Scheme 28 | |
9
endo-Cyclizations
The 5-endo cyclization of allenes with a nucleophilic group in the α-position is now well established, since the accidental re-discovery of Olsson and Claesson's reaction23 by Marshall using rhodium(I) and silver. Three groups are generally used: alcohols leading to dihydrofurans, ketones leading to furans, and carboxylic acids leading to butenolides (Scheme 29). A reasonable mechanism would involve metal co-ordination to one allenic double bond, followed by nucleophilic attack to generate a metalloheterocycle (18) (Scheme 30). Subsequent protonolysis of the carbon–metal bond would then yield the product and regenerate the catalyst. Six-membered rings may also be formed.23
 |
| | Scheme 29 | |
 |
| | Scheme 30 | |
Examples of the latter two processes are both from Marshall's synthesis of Kallolide A (19).33
Treatment of allenic ketone (20) with a catalytic amount of silver nitrate in refluxing acetone yielded the required furan (21) (Scheme 31).
 |
| | Scheme 31 | |
To generate the butenolide (22), allenic acid (23) was treated under very similar conditions. A small amount of a diastereomer was also formed (Scheme 32).
 |
| | Scheme 32 | |
Standaert and Van Brundt have recently used the allenic alcohol endo-cyclization in a synthesis of furanomycin (24) (Scheme 33).34
 |
| | Scheme 33 | |
While Marshall has employed both rhodium(I) and silver(I), Hashmi has examined a range of metals for activity in the furan forming reaction.35 Copper(I), silver(I) and rhodium(II) proved to be effective, but slow. Ruthenium(II) and palladium(II) resulted in faster reactions. Subsequently, he found that gold(III), previously used in alkyne cyclizations, is an even faster catalyst36
However, with gold and palladium substantial amounts of dimeric products were formed when monosubstituted allenes were the substrates (Scheme 34). In the case of palladium, the 3-palladiofuran intermediate was trapped by a second molecule of allene faster than it under went protonolysis, leading to the 3-substituted furan (25) as the only product.
 |
| | Scheme 34 | |
In contrast, with gold(III), the second molecule of allene became attached at the 2-position (Scheme 35). A reasonable explanation is that the 3-auriofuran (26) undergoes protonolysis to liberate Au(III) which then counterattacks at the 2-position in an electrophilic aromatic substitution reaction.
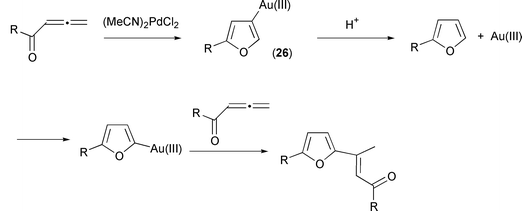 |
| | Scheme 35 | |
A corollary of this mechanism is that free furans should react with Michael acceptors in the presence of a gold(III) catalyst, in an alternative to cuprate additions (Scheme 36). Hashmi has shown that this is indeed so.
 |
| | Scheme 36 | |
The gold catalyzed cyclization also works for the formation of dihydrofurans from α-hydroxyallenes (Scheme 37).37 Although this reaction can be achieved with just anhydrous acids, the advantage of gold is that acid sensitive functional groups, such as silyl ethers, survive. The axial stereochemistry of the allene is efficiently relayed to the dihydrofuran.
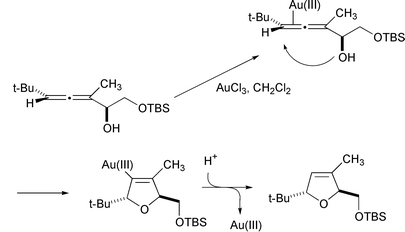 |
| | Scheme 37 | |
endo-Cyclizations have also been achieved using organo-palladium(II) species generated through oxidative addition (Scheme 38). These have been employed to form β-arylbutenolides,38 3-acylpyrrolidines15 and other heterocycles. A number of other examples of endo-cyclizations have been mentioned elsewhere in this review (Schemes 6, 7, 18, 26).
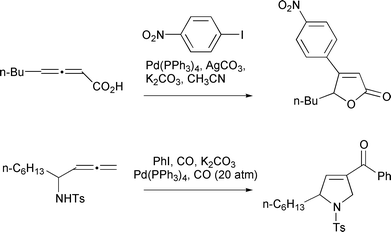 |
| | Scheme 38 | |
A striking extension of this process is the cyclization of a 1,2,3-triene, generated in situ, to a tetrasubstituted furan (Scheme 39).39
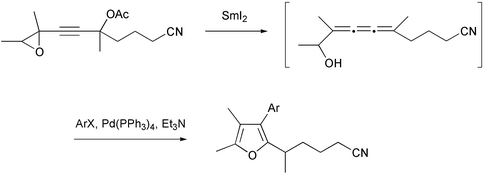 |
| | Scheme 39 | |
10 Diastereoselectivity
In reactions leading to five-membered rings, a substituent α-to the allene exerts a powerful influence in favour of the trans-product (Scheme 40). This is true for several cyclization reactions and metals.17,18,24,40
 |
| | Scheme 40 | |
In contrast, the few examples with a substituent β- to the allene result in poor diastereoselectivity.17 A γ-substituent (i.e.
α to the nucleophilic group), gives variable results in favour of either the cis or trans products depending on the nucleophile and the catalyst system (Scheme 41).4,14,17,26,28 In one case, using a silver catalyzed cyclization, the high stereoselectivity of the reaction allowed a formal synthesis of α-anatoxin (27).41
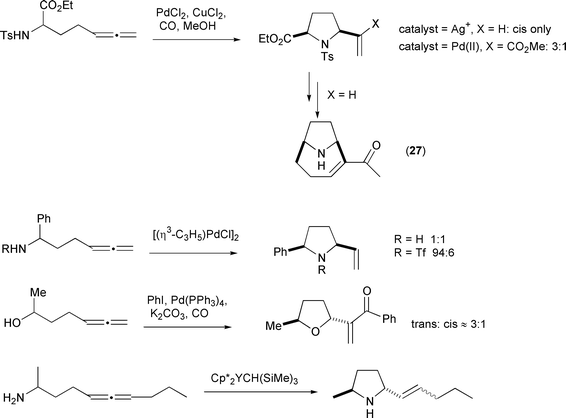 |
| | Scheme 41 | |
In the six-membered ring series, it is notable that the few reported examples involve the formation of diequatorial products as the major product.6,20,26 Examples have been shown in Schemes 21 and 14. Variable diastereoselectivity has been observed in small ring formation.12,13
The influence of chirality outside of the ring being formed has received little attention. Gallagher has reported the silver mediated cyclization of amino allenes bearing chiral auxiliaries on the nitrogen nucleophile.42 Auxiliaries with a group capable of coordination gave the highest diastereoselectivites. Curiously, the results were dependent on the amount of catalyst employed indicating that the stereochemistry-determining step is not a simple 1 ∶ 1 allene–metal complex. The same substrates, under palladium catalyzed carbonylation conditions gave very modest diastereoselectivities. In addition, the use of chiral ligands for the palladium had only ‘marginal’ effects. Despite this, the diastereoisomeric products are frequently capable of quite facile chromatographic separation. The enantiomerically pure products are therefore available through this route, albeit by a kind of resolution procedure. In this way, the allene (28)
could be cyclized, separated and carried on to Pumiliotoxin 251D (29) (Scheme 42).43
 |
| | Scheme 42 | |
11 Ring sizes
The most common application of this chemistry, not surprisingly, is to the formation of five membered rings by an exo cyclization process. A large number of six membered rings have also been formed. Given the strain involved, the recent reports of the formation of three and four membered rings are highly interesting. Such heterocycles are, themselves, reactive with low-valent palladium and their isolation is clearly due to a judicious or fortuitous choice of conditions and to the kinetic barriers to their conversion to the isomeric 5 or 6 membered rings.
Few attempts at larger rings have been reported. Apart from Trost's macrocyclization,30 the application of Gallagher's nitrone strategy is notable.44 Kang has been able to use aryl palladium(II) triggers, derived from iodonium salts, to generate a nine and a ten membered ring (Scheme 43)8 and Takahashi has been able to generate seven and eight membered ring lactones.31
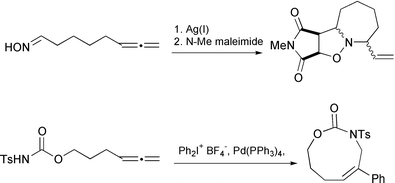 |
| | Scheme 43 | |
12 Conclusion
This is a fast growing area of organic chemistry. From these allenes, a wide range of products can be formed, often with good stereo- and regio-chemical control. The use of ‘less common’ metals such as the lanthanides and gold is a sure indicator that new developments will be forthcoming. Further applications of these reactions in total syntheses can also be anticipated.
13 Acknowledgements
Our own work in this area is generously supported by the Thailand Research Fund.
14 References
- M. Frederickson and R. Grigg, Org. Prep. Proced. Int., 1997, 29, 65 Search PubMed
 .
.
-
H. F. Schuster and G. M. Coppola, Allenes in Organic Synthesis, Wiley, New York, 1984 Search PubMed .
- For a wide ranging review of Pd catalyzed allene reactions of all types, see R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 Search PubMed .
- I. W. Davies, D. I. C. Scopes and T. Gallagher, Synlett, 1993, 85 CrossRef CAS .
- W. F. J. Karstens, D. Klomp, F. P. J. T. Rutjes and H. Hiemstra, Tetrahedron, 2001, 57, 5123 CrossRef CAS .
- M. Kimura, S. Tanaka and Y. Tamaru, J. Org. Chem., 1995, 60, 3764 CrossRef CAS .
- R. D. Walkup, L. Guan, M. D. Mosher, S. W. Kim and Y. S. Kim, Synlett, 1993, 88 CrossRef CAS .
- S.-K. Kang, T.-G. Baik and Y. Hur, Tetrahedron, 1999, 55, 6863 CrossRef CAS .
- W. F. J. Karstens, M. Stol, F. P. J. Rutjes and H. Hiemstra, Synlett, 1998, 1126 CrossRef CAS .
- S.-K. Kang, T.-G. Baik and A. N. Kulak, Synlett, 1999, 324 CAS .
- F. P. J. T. Rutjes, K. C. M. F. Tjen, L. B. Wolf, W. F. J. Karstens, H. E. Schoemaker and H. Hiemstra, Org. Lett., 1999, 1, 717 CrossRef CAS .
- S. Ma and S. Zhao, J. Am. Chem. Soc., 1999, 121, 7943 CrossRef CAS .
- H. Ohno, M. Anzai, A. Toda, S. Ohishi, N. Fujii, T. Tanaka, Y. Takemoto and T. Ibuka, J. Org. Chem., 2001, 66, 4904 CrossRef CAS .
- R. D. Walkup, L. Guan, Y. S. Kim and S. W. Kim, Tetrahedron Lett., 1995, 36, 3805 CrossRef CAS .
- S. K. Kang and K. J. Kim, Org. Lett., 2001, 3, 511 CrossRef CAS .
- C. Jonasson, A. Horváth and J.-E. Bäckvall, J. Am. Chem. Soc., 2000, 122, 9600 CrossRef CAS .
- T. Gallagher, I. W. Davies, S. W. Jones, D. Lathbury, M. F. Mahon, K. C. Molloy, R. W. Shaw and P. Vernon, J. Chem. Soc, Perkin Trans. 1, 1992, 433 RSC .
- R. D. Walkup and M. D. Mosher, Tetrahedron
Lett., 1994, 35, 8545 CrossRef CAS .
- R. D. Walkup and G. Park, J. Am. Chem. Soc., 1990, 112, 1597 CrossRef CAS .
- B. B. Snider and F. He, Tetrahedron Lett., 1997, 38, 5453 CrossRef CAS .
- R. W. Bates, T. Rama-Devi and H.-H. Ko, Tetrahedron, 1995, 51, 12939 CrossRef CAS .
- B. M. Trost, A. B. Pinkerton and D. Kremzow, J. Am. Chem. Soc., 2000, 122, 12007 CrossRef CAS .
- L.-I. Olsson and A. Claesson, Synthesis, 1979, 743 CrossRef CAS .
- M. Kimura, K. Fugami, S. Tanaka and Y. Tamaru, Tetrahedron Lett., 1991, 32, 6359 CrossRef CAS .
- D. C. Lathbury, R. W. Shaw, P. A. Bates, M. B. Hursthouse and T. Gallagher, J. Chem. Soc., Perkin Trans. 1, 1989, 2415 RSC .
- V. M. Arredondo, S. Tian, F. E. McDonald and T. J. Marks, J. Am. Chem. Soc., 1999, 121, 3633 CrossRef CAS .
- S. Tian, V. M. Arredondo, C. L. Stern and T. J. Marks, Organometallics, 1999, 18, 2568 CrossRef CAS .
- M. Meguro and Y. Yamamoto, Tetrahedron Lett., 1998, 39, 5421 CrossRef CAS .
- S. Kamijo and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 1747 CrossRef CAS .
- B. M. Trost, P.-Y. Michellys and V. J. Gerusz, Angew. Chem., Int. Ed., Engl., 1997, 36, 1750 CrossRef CAS .
- E. Yoneda, S.-W. Zhang, K. Onitsuka and S. Takahashi, Tetrahedron Lett., 2001, 42, 5459 CrossRef CAS .
- S. K. Kang, K. J. Kim, C.-M. Yu, J.-W. Hwang and Y.-K. Do, Org. Lett., 2001, 3, 2851 CrossRef CAS .
- J. A. Marshall and J. Liao, J. Org. Chem., 1998, 63, 5962 CrossRef CAS .
- M. P. Van Brundt and R. F. Standaert, Org. Lett., 2000, 2, 705 CrossRef .
- A. S. K. Hashmi, Angew. Chem., Int. Ed. Engl., 1995, 34, 1581 CrossRef .
- A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285 CrossRef CAS .
- A. Hoffman-Röder and N. Krause, Org. Lett., 2001, 3, 2537 CrossRef .
- S. Ma and Z. Shi, J. Org. Chem., 1998, 63, 6387 CrossRef CAS .
- J. M. Aurrecoechea and E. Pérez, Tetrahedron Lett., 2001, 42, 3839 CrossRef CAS .
- R. W. Bates and V. Satcharoen, Synlett, 2001, 532 CAS .
- N. J. S. Huby, R. G. Kinsman, D. Lathbury, P. G. Vernon and T. Gallagher, J. Chem. Soc., Perkin Trans. 1, 1991, 145 RSC .
- D. N. A. Fox and T. Gallagher, Tetrahedron, 1990, 46, 4697 CrossRef CAS .
- D. N. A. Fox, D. Lathbury, M. F. Mahon, K. C. Molloy and T. Gallagher, J. Am. Chem. Soc., 1991, 113, 2652 CrossRef CAS .
- R. Shaw, D. Lathbury, M. Anderson and T. Gallagher, J. Chem. Soc.,
Perkin Trans. 1, 1991, 659 RSC .
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here. 