DOI:
10.1039/B107811A
(Review Article)
Chem. Soc. Rev., 2002,
31, 1-11
Atom transfer radical cyclisation reactions mediated by copper complexes
Received
1st September 2001
First published on 3rd December 2001
Abstract
This article describes recent advances in the use of copper complexes in mediating atom transfer radical cyclisation reactions (ATRC). Recent developments have included the design of activated complexes which mediate the cyclisation of tri-, di-, and mono-halo derived substrates at ambient temperatures. Using this methodology, cyclisation to give a variety of ring sizes (4–18 membered rings) has been demonstrated. In addition tandem and radical–polar crossover reactions have also been developed. The design of solid supported and perfluorous complexes that mediate cyclisations may make this approach to the synthesis of rings more attractive towards industrial applications.
 Andrew J. Clark Andrew J. Clark |
Dr Andrew Clark is a Senior Lecturer in Synthetic Chemistry at the University of Warwick (UK). He obtained his PhD from King’s College, London (1990) working in the area of ‘organocobalt mediated free radical reactions’ under the supervision of Professor K. Jones. After postdoctoral work in the groups of Professor Gilbert Stork (Columbia University, New York, 1992) and Professor Gerald Pattenden (University of Nottingham, 1993) he became a lecturer at the University of Warwick (1993). His research interests include free radical chemistry, total synthesis of natural products and the use of renewable resources as chemical feedstocks for the polymer industry. |
1 Introduction
The efficient preparation of cyclic systems continues to be an important area of modern organic chemistry. The formation of such cyclic systems by carbon–carbon bond formation has increasingly been achieved by the use of free radical cyclisation protocols.1 The majority of such reactions are mediated by organostannane or organosilane reagents (e.g. Bu3SnH or HSi(SiMe3)3) (Scheme 1).1
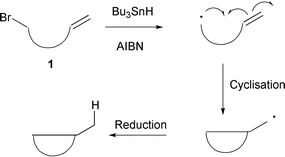 |
| | Scheme 1
Reductive cyclisations using Bu3SnH.
| |
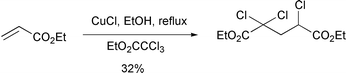
The disadvantage of these methods is that they are reductive in nature, i.e. the cyclised radicals are ‘quenched’ by the addition of a hydrogen atom from the mediating reagent. This approach can be limited as it leads to the loss of two functional groups. For example, cyclisation of 1 with Bu3SnH leads to the loss of both the radical precursor (halogen) and the radical acceptor (alkene). In addition, the use of organostannane reagents is complicated by their high toxicity, high expense and purification problems. Transition metal catalysed intermolecular addition of polyhalocarbon derived molecules to alkenes has been known for some time (Scheme 2).2 The intramolecular version of this reaction, atom transfer radical cyclisation (ATRC), can provide a convenient method for the construction of various ring systems. In particular ATRC reactions of 2,2,2-trichlorinated carbonyl compounds
have been reported with a range of metal catalysts, e.g. Ni metal,3 RuCl2(PPh3)3, and FeCl2(P(OEt)3)3.4 However, by far the most successful catalysts have been those derived from copper(I)-based halogen compounds.4 These oxidative atom transfer cyclisations involve redox reactions between copper(I) and copper(II) complexes. Thus, reaction of an activated trichloroacetate such as 2 with CuCl in MeCN at 140 °C in a pressure bottle for 1 hour generates the initial radical 3 and CuCl2 (Scheme 3).5 After cyclisation the newly formed (more reactive) primary radical 4 reacts with CuCl2 to regenerate the CuCl catalyst and furnish the cyclised product 5. The use of copper complexes
in mediating radical cyclisations thus has a number of advantages over alternative reductive methods including: (a) the low cost of copper halides, (b) the ease of work-up of the reactions, and (c) the catalytic nature of the processes.
 |
| | Scheme 2 | |
 |
| | Scheme 3
Oxidative copper(I) mediated atom transfer radical cyclisation.
| |
2 Reactions mediated by CuCl and CuCl–bipyridine complex
2.1 Reactions using CuCl
Atom transfer radical cyclisation mediated by catalytic amounts of CuCl has been utilised to prepare not only γ-lactones but also γ-lactams.6,7 Heating both the trichloroacetamide derivatives 6a,b with CuCl in MeCN at 140 °C furnished the desired 5-exo atom transfer products 7a,b in 57% and 87% yield respectively (Scheme 4).6 No products arising from 6-endo cyclisation were detected. Using this protocol it was possible to cyclise both secondary 6a and tertiary 6b amides, although the cyclisation of the tertiary amide 6b was the more efficient of the two. Bicyclic lactams were also readily prepared using this approach providing access to pyrrolidine alkaloid skeletons (e.g. 9). Cyclisation of 8 at 110 °C furnished one diastereoisomer 9 containing the cis fused
ring junction in 91% yield (Scheme 5).7
 |
| | Scheme 4 | |
 |
| | Scheme 5 | |
The cyclisation of trichloroacetates derived from secondary alcohols 10a,b proceeds with excellent diastereoselectivity to furnish predominantly the trans diastereomers 11a,b (Scheme 6).8 Interestingly the stereochemical outcome of the cyclisation reactions of 10 was similar to those obtained using other free radical processes.
 |
| | Scheme 6 | |
2.2 Effect of copper salt, solvent and other additives
Screening of other copper salts indicated that a range were effective in mediating the cyclisation of trichloroacetate 2 at elevated temperatures including Cu2O, Cu(NO3)2·H2O, and Cu(CCPh).8 The concentration of the reactions was also found to be crucial, with relatively high concentrations leading to telomerisation of the substrate. While a range of solvents was investigated, only acetonitrile and alcohols were found to be effective in mediating the cyclisation of trichloroacetate 2.8 However, the addition of an equimolar amount of 2,2′-bipyridine (bipy) to CuCl was found to accelarate the rate of the reaction fourfold (Scheme 7).8 In general, the addition of either amine or pyridine ligands to atom transfer reactions has been found to cause rapid rate accelerations for a variety
of cyclisation and intermolecular addition reactions (see Sections 4.1–4.4).
 |
| | Scheme 7 | |
2.3 Use of copper halide–2,2′-bipyridine complexes
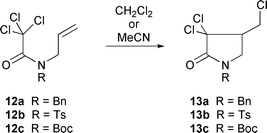
Ligands may act to accelerate atom transfer processes by solubilising the CuCl, or by altering the redox potential of the catalyst system, or by both. Screening the substrates 12a–c with various ligand systems indicated that the use of 30 mol% of a 1 ∶ 1 mixture of CuCl–bipy in CH2Cl2 catalysed the cyclisation more rapidly than CuCl in MeCN.9 Other solvents such as 1,2-dichloroethane and THF were also compatible with the use of CuCl–bipy as a mediator. Thus, with this more activated catalyst system it was possible to cyclise a variety of substrates at room temperature or below. The nature of the N-protecting group was also found to affect the rate of the cyclisation (Table 1).
| Substrate |
Catalyst (mol%)a |
Temp./°C |
Time/h |
Yield (%) |
|
Reactions with CuCl were run in MeCN, reactions with CuCl–bipy were run in CH2Cl2.
|
|
12a
|
CuCl (30) |
80 |
18 |
68 |
|
12a
|
CuCl–bipy (30) |
rt |
1 |
98 |
|
12b
|
CuCl (30) |
rt |
24 |
97 |
|
12b
|
CuCl–bipy (5) |
rt |
0.2 |
91 |
|
12c
|
CuCl (30) |
80 |
4 |
80 |
|
12c
|
CuCl–bipy (30) |
rt |
2 |
78 |
In general the cyclisation of α-N-allylcarbamoyl radicals (derived from 12) is a difficult process requiring high temperatures, primarily due to the high barrier to rotation which characterises the amide bond. Only one conformer can cyclise (anti) and the nature of the N-protecting group alters the conformer population, thus bulky or electron withdrawing substituents (12b,c) favour cyclisation by shifting the equilibrium towards the anti conformer (Scheme 8).10
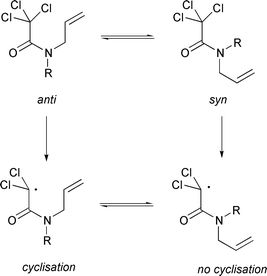 |
| | Scheme 8 | |
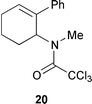
This phenomenon has been used to good effect in the formal total synthesis of both mesembrane 14 and crinane 15.10 Cyclisation of 16a,b with 30 mol% CuCl–bipy furnished the products 17a,b in 78% and 78% yield respectively (Scheme 9). Manipulation of these intermediates to the dl-natural products, mesembrane 14 and crinane 15, was then accomplished using standard chemistry.10 Attempts to mediate the cyclisation of 18 which contained the desired N-Me group only produced low yields (20%) of the desired product 19 (Scheme 10) due to population of rotamers unfavourable to cyclisation (e.g.20).10
 |
| | Scheme 9 | |
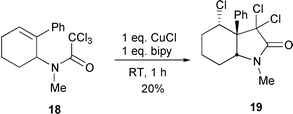 |
| | Scheme 10 | |
The stereochemical outcome of the 5-exo cyclisation of simple precursors was also found to be N-protecting group dependent with N-benzyl compound 21a leading to the trans isomer 22a, while the N-tosyl substrate 21b led to the cis product 23b predominantly (Scheme 11).9 The electron withdrawing N-tosyl group has also been used to facilitate the cyclisation of various N-allylhalodifluoroacetamides 24 to give α,α-difluorinated-γ-lactams 25 (Scheme 12). The reactivity of halodifluoroacetamides was found to decrease in the order I > Br >> Cl (using CuI, CuBr and CuCl–bipy mixtures respectively).11
 |
| | Scheme 11 | |
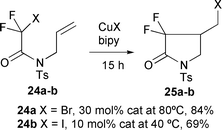 |
| | Scheme 12 | |
The highly activated nature of the CuCl–bipy catalyst system allows the cyclisation of mono-halo substrates at elevated temperatures (80 °C). Hence, application of this protocol to the synthesis of cyclic amino acids has also been reported.12 Best results were obtained when the reactions were performed at 80 °C for 18 hours (Scheme 13). A variety of solvents were found to be compatible with the cyclisation of the glycine derived radicals, however the use of good hydrogen atom donors like THF, dimethoxyethane and acetone led to substantial amounts of reduced cyclisation products. The regioselectivity and stereoselectivity of the cyclisations were found to parallel those obtained from Bu3SnH mediated cyclisations. In analogous chemistry the cyclisation of radicals derived from 2-(alk-3-en-1-oxy)-2-chloroacetates was possible (Scheme 14).13
Interestingly the regioselectivity of the reactions was dependent upon the copper complex used. Hence, cyclisation of 26 with CuCl–2,2′-bipyridine proceeded as expected to give the 5-exo product 27, while the use of 6,6′-bipyridines led exclusively to the 6-endo product 28.13 This was rationalised as being due to the different ligands promoting either a radical or cationic cyclisation pathway respectively. The use of CuCl–bipy in the cyclisation of a range of 2-(alk-3-en-1-oxy)-2-chloroacetates furnished 3-(1-chloroalkyl)-substituted tetrahydrofurans in good yields. Hence, cyclisation of 29 furnished 30 in 95% yield as an 82 ∶ 18 mixtures of isomers respectively (Scheme 15). The chlorine substituent incorporated in the products could be used to good effect in further chemistry (e.g. lactonisation reactions). The utilisation
of this atom transfer–lactonisation protocol allowed for the efficient total synthesis of the natural products avenaciolide and isoavenaciolide.13
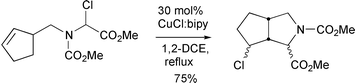 |
| | Scheme 13 | |
 |
| | Scheme 14 | |
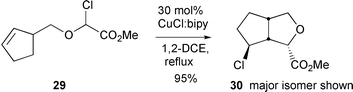 |
| | Scheme 15 | |
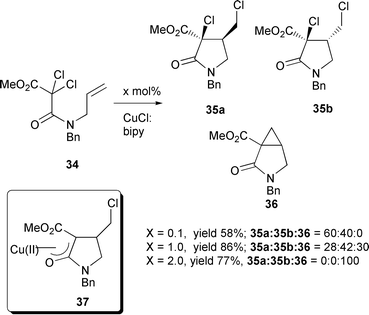
A 1 ∶ 1 mixture of CuCl–bipy has also been used in the synthesis of 2,3-disubstituted 4-chlorofurans.14 Hence, reaction of 1-acetoxy-2,2,2-trichloroethyl allyl esters 31 with 30 mol% CuCl–bipy in refluxing 1,2-dichloroethane (DCE) furnished the polychlorinated tetrahydrofurans 32. Dechloroacetoxylation with Zn dust followed by tandem dehydrohalogenation–aromatisation with t-BuOK and 18-crown-6 led to the desired furan derivatives 33 (Scheme 16).14 The reaction of dichloromalonic derivatives, where the halogens are adjacent to two carbonyl groups, leads to a variety of products depending upon the relative amount of CuCl–bipy used (Scheme 17.15 For example with 10 mol% only the two diastereomers 35a,b arising from 5-exo cyclisation were detected
(in a 6 ∶ 4 ratio), however when a stoichiometric amount of copper complex was utilised the bicyclic cyclopropane product 36 was also detected and the ratio of 35a,b had reversed.15 The use of two equivalents of CuCl–bipy led to exclusive formation of the 3-azabicyclo[3.1.0]hexan-2-one 36. This was postulated to arise via a sequential radical cyclisation followed by an intramolecular nucleophilic substitution reaction by the Cu(II) enolate 37.15
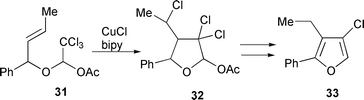 |
| | Scheme 16 | |
 |
| | Scheme 17 | |
3 Medium ring heterocycles
The synthesis of medium ring lactones and lactams (8–12 membered rings) continues to be an active area of research due to the large number of natural products that contain these frameworks. Although there is a range of methods to prepare such systems, the use of radical chemistry to facilitate macrocyclisations is less developed. Medium-sized lactones can be efficiently prepared by the reaction of di- and trichloroacetates using CuCl–bipy (Scheme 18).16 Reaction of compounds 38a–d in the presence of 30 mol% CuCl–bipy in a 0.1 M solution of refluxing 1,2-dichloroethane for 18 hours furnished the desired products 39a–d in various yields (57–75%). The endo mode of cyclisation was exclusively observed and corresponds to that predicted by calculations and other reported radical processes.16 This is primarily observed in systems
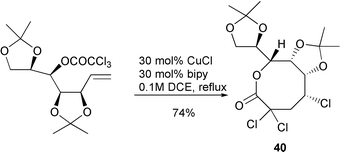
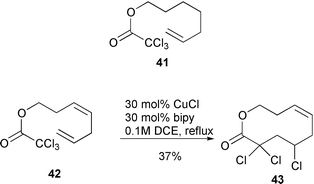
which contain a relatively long chain due to the fact that the most stable conformation for the ester function (s-trans) does not impede the cyclisation process. The reactions were found to proceed in various solvents including refluxing benzene. A wide range of 8-membered ring systems have been prepared via this approach, including the highly functionalised lactone 40 in 74% yield as a single diastereomer (Scheme 19).17 While it was possible to prepare 8- and 9-membered rings in good yield via this protocol it was not possible to prepare larger ring sizes, unless a rigid element of functionality was built into the cyclisation precursor. Thus, while cyclisation of 41 failed with only telomers being detected, the related system 42 successfully gave the 10-membered lactone 43 in 37% yield after three days at reflux (Scheme 20).18 Presumably the rigid element incorporated in the carbon chain promotes intramolecular addition via entropy effects.
 |
| | Scheme 18 | |
 |
| | Scheme 19 | |
 |
| | Scheme 20 | |
It was found that 0.3 equivalents of CuCl–bipy was essential for all the macrocyclisations reported above. If less catalyst was used lower yields resulted due to catalyst decomposition before completion of the reaction. It was also found crucial to facilitate these reactions using concentrations between 0.07–0.02 M in solution to avoid telomerisation processes.18 The high efficiency of these medium ring cyclisations coupled with the fact that it was impossible to cyclise 38c using reductive Bu3SnH technology has prompted some researchers to speculate that the reactions may be proceeding via metal co-ordinated radicals or that cyclisation is taking place within the co-ordination sphere of the copper complex leading to a templating process.18 Despite the versatility of this procedure for the preparation of 8- and 9-membered rings, the inability to cyclise larger systems without rigid
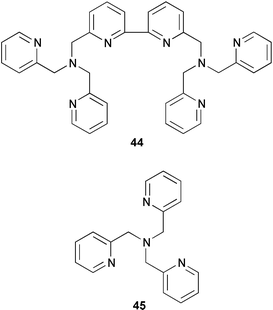
control elements, and the need for relatively large amounts of catalyst (0.3 equivalents) to facilitate the reactions is a distinct disadvantage. This has prompted a number of researchers to investigate a range of alternative copper complexes to mediate ATRC reactions. Recently, Verlhac et al. reported that the multidentate pyridine ligands 44 and 45 were superior to bipyridine in a range of macrocyclisation reactions.19
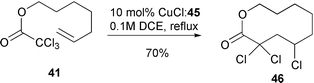
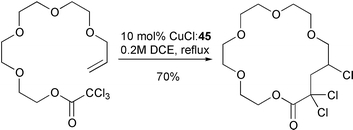
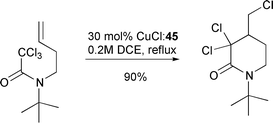
Thus, cyclisation of 38a could be achieved in 99% yield with 0.1 equivalent of catalyst derived from ligand 44 (only 47% yield was obtained under identical conditions with bipyridine as ligand). Even more impressive was that it was possible to produce 39a in 90% yield with only 0.03 equivalents of the catalyst derived from the more active tripyridylamine ligand 45. In fact, the use of this ligand system enabled macrocyclisations of substrates which were impossible using bipyridine as ligand (Scheme 21)19 (e.g. heating 41 with 0.1 equivalent of CuCl–45 furnished 46 in 70% yield whereas only telomers were isolated when CuCl–bipy was utilised). A range of crown ethers, Scheme 22)20 and δ-lactams, (Scheme 23) have also been prepared
using this approach.21
 |
| | Scheme 21 | |
 |
| | Scheme 22 | |
 |
| | Scheme 23 | |
4 Reactions mediated by other CuCl and CuBr complexes
4.1 Second generation of copper catalysts and structure–activity relationships
The discovery that different ligands can modify the reactivity, yield and selectivity of atom transfer reactions has prompted various groups to investigate the use of alternative ligand systems in cyclisation reactions. The ability to fine-tune both the solubility and redox potential of the catalysts by varying the ligand has enabled a range of highly activated catalyst systems to be prepared. In addition the choice of ligand used can often modify the product distribution significantly (see Table 2). By far the most useful of the new more active generation of atom transfer catalysts are those based upon a) N-alkyl-2-pyridylmethanimines (NPMI) 47,21–26 b) N,N,N′,N’-tetramethylethylenediamine (TMEDA) 48,27–30 and c) N,N,N′,N′,N″,N″-hexamethyltriethylenetetramine
(Me6-tren) 49.21,25,26,31
| Ligand |
R group |
Relative ratea |
dr |
|
Relative rate with respect to the reaction of ligand 47d.
|
|
47a
|
n-Bu |
45 |
18 ∶ 82 |
|
47b
|
i-Bu |
28 |
28 ∶ 72 |
|
47c
|
s-Bu |
3 |
32 ∶ 68 |
|
47d
|
t-Bu |
1 |
51 ∶ 49 |
4.2
N-Alkyl-2-pyridylmethanimines (NPMI’s)
In systems where copper halides are used in conjunction with bipy, the bipy is thought to primarily serve to solubilise the copper halide as [Cu(I)(bipy)2]X. In addition, the low lying LUMO π* orbital, present in the conjugated π-system of bipy, is able to accept electron density from the metal and hence serve to stabilise the Cu(I) oxidation state. The structurally similar NPMI ligands have also been reported to solubilise Cu(I) halides and also have low lying π* orbitals.22 However, the relative ease of preparation of these ligands (easily prepared by reaction of commercially available amines with pyridine carbaldehydes in the presence of MgSO4) has allowed a whole range of catalysts with different solubilities, steric and electronic structures to be prepared.23 Structure–activity relationships have indicated that the nature of the
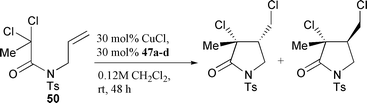
imine substituent is crucial in controlling the rate and selectivity of the cyclisation reaction. Hence, cyclisation of 50 with the range of ligands shown indicates that bulky substituents retarded the rate of cyclisation significantly (Table 2).23 In addition the diastereoselectivity of the process was also affected by the nature of the N-alkyl group. The optimum ratio of ligand to copper halide was found to be 2 ∶ 1.22,23
The solubility of the complexes could be altered by tailoring the length of the N-alkyl substituent (e.g., 47e R = n-Pr the catalyst was soluble in water at room temperature, insoluble in toluene at room temperature but soluble in toluene at 110 °C). The active nature of the catalyst 47f (R = n-pentyl) allowed for the cyclisation of mono-halosubstrates such as 51 at room temperature.23 Screening of a range of electronically modified ligands 52a–d in the cyclisation of 51 (Table 3) indicated that the order of reactivity was 52b>52a>52c>52d (paralleling σm in relation to the pyridine nitrogen). This suggests that inductive effects onto the pyridine nitrogen and not resonance effects onto the imine nitrogen are the dominant features for this class of ligand in cyclisation
reactions.24 The ligand 52b which contained a mildly inductive electron donating group (which causes an increase in the energy of the ligand LUMO and thus a decrease in the relative stability of the Cu(I) oxidation state) showed a rate enhancement, whereas those with electron withdrawing inductive groups caused a decrease in the rate of reaction. Thus the rates of ATRC reactions may well parallel the basicity of the pyridine nitrogen.24
| Ligand |
R group |
Ratio 51
∶
53 |
|
52a
|
H |
41 ∶ 69 |
|
52b
|
Me |
34 ∶ 66 |
|
52c
|
OMe |
73 ∶ 27 |
|
52d
|
NO2 |
>98 ∶ 2 |
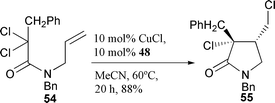
One of the major disadvatages of the CuCl–bipy reagent system is that substantial amounts of catalyst (normally 30 mol%) of this relatively expensive reagent are required for efficient catalysis. The use of the more reactive CuCl–TMEDA reagent combination furnishes a catalyst system which gives better yields at lower catalyst loading in simple ATRC reactions. In addition, cyclisations can often be carried at out at lower temperatures than with CuCl–bipy. An added advantage is the relative inexpense of the TMEDA additive. As in the case of the bidentate NMPI ligands, the optimum ratio of the bidentate TMEDA ligand to CuCl was found to be 2 ∶ 1.27 Thus CuCl–(TMEDA)2 complex can mediate efficient 5-exo cyclisations of a range of trichloro and dichloracetamide derivatives. Cyclisation of 54 with 10 mol% CuCl–(TMEDA)2 in acetonitrile for 20 hours at 60 °C furnished the product 55
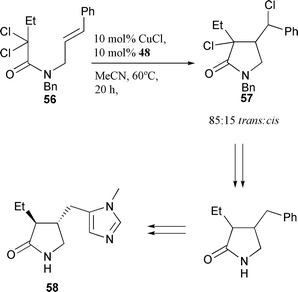
as one diastereomer in 88% yield (Scheme 24).27 Interestingly, attempts to cyclise this substrate with CuCl–bipy failed, indicating the importance of utilising the correct choice of ligand for a given cyclisation. The influence of the N-benzylic protection in the CuCl–(TMEDA)2 catalysed 5-exo cyclisation of a range of chiral substrates has been examined.28,29 The steric nature of the N-substituent was not found to influence the stereoselectivity of the cyclisations to any significant extent.28 The synthetic utility of CuCl–(TMEDA)2 promoted cyclisation has been explored by application to the formal total synthesis of pilolactam 58 (Scheme 25)28 and in the synthesis of 3-benzyliminopyrrolidin-2-ones.30
 |
| | Scheme 24 | |
 |
| | Scheme 25 | |
4.4
N,N,N′,N′,N″,N″-Hexamethyltriethylenetetramine (Me6-tren)
The origin of the reported improvement in the activity of CuCl(TMEDA)2 relative to CuCl(bipy) has been speculated to arise due to the fact that simple copper(amine) complexes have lower redox potentials than copper(pyridine) complexes. Ghelfi and co-workers reported that the optimium ratio for copper halide–TMEDA was 1 ∶ 2 indicating that two equivalents of bidentate ligand are required to make the active catalyst.27 As a consequence of this observation a range of other multidentate amine ligands have also been screened in ATRC reactions. The most active polydentate amine ligand found to date is the tetradentate Me6-tren ligand 49.21,31 The use of a 1 ∶ 1 ratio of copper halide:49 in various solvents was found to produce a catalyst far more active than either bipyridine, NMPI or TMEDA. Thus cyclisation of 59 proceeded only slowly at room temperature
with NMPI 52 (72 h, 15% conversion) but rapidly (less than 2 h, yield 90%) with Me6-tren 49 (Scheme 26). The more activated nature of the catalyst allowed cyclisation to take place with lower catalyst loadings, thus cyclisation of 50 was accomplished with only 5 mol% catalyst at room temperature in 24 hours.21,31 While it was possible to use even lower catalyst loadings at room temperature (e.g. 0.5 mol%) the reaction only proceeded to give a 33% conversion in the same 24 hour period. Cyclisation of a range of monohalosubstrates 60a,b was possible at room temperature (Scheme 27). Cyclisation of the primary bromide 60c was possible, albeit at elevated temperature. The product was obtained in low yield due to competing amide cleavage.21
 |
| | Scheme 26 | |
 |
| | Scheme 27 | |
Using this protocol it was not necessary to use vigorously dried glassware or solvents. In addition work-up of the reactions was facile, as the crude reaction mixture was passed through a small silica plug and the solvent removed to furnish the atom transfer products directly. Attempts to mediate 8-endo macrocyclisations of N-tosylamide 61 using this ligand system failed, with the main products being the rearranged compound 62 as well as unreacted starting material 61.21 The rearranged product 62 was postulated to arise via a competing 5-exo ipso aromatic radical substitution to give 63 followed by re-aromatisation followed by C–S bond cleavage to give 64, loss of SO2 and reduction of the resulting amide radical (Scheme 28).
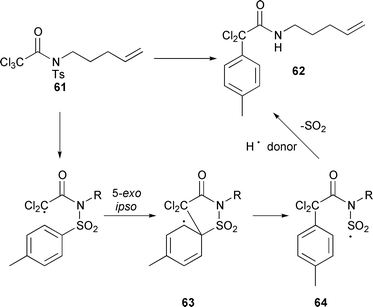 |
| | Scheme 28 | |
5 Sequential reactions
The ability of copper(I) complexes to catalyse both cyclisation (ATRC) and intermolecular addition (ATRA) reactions has been exploited in a variety of tandem sequences. Intermolecular addition reactions are generally slower than intramolecular processes and this phenomenon has been used in sequential reactions. Thus, heating trichloroacetamide 12b with methylenecyclohexane 65 with 10 mol% CuCl–bipy at 83 °C for 1 hour furnished the functionalised product 66 in 85% yield as almost a single diastereomer (Scheme 29).32 In this case after cyclisation, the activated nature of the α,α-dichlorocarbonyl functionality in 13b allows for the generation of a new cyclic radical 67. This then undergoes an intermolecular addition reaction and is trapped by atom transfer from the CuCl2–bipy generated in the second initiation step.32
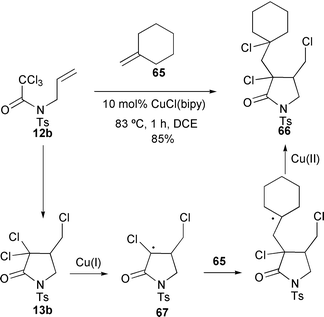 |
| | Scheme 29 | |
This approach has also been used to facilitate tandem cyclisations. Thus reacting 68 at room temperature with 30 mol% CuCl–bipy at 40 °C furnished the two diastereomers 70 in 83% yield. Purification using silica chromatography furnished the corresponding exocyclic alkenes 71 after elimination of HCl (Scheme 30).32
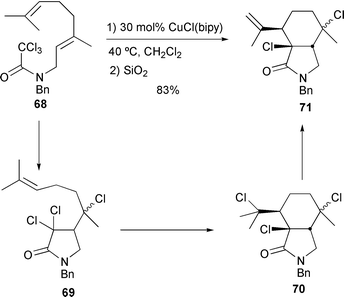 |
| | Scheme 30 | |
6 Solid supported ATRC catalysts
In order to facilitate work-up of atom transfer reactions as well as develop re-usable catalysts the immobilisation of pyridine–imine ligands onto solid supports has been investigated. Reaction of pyridine-2-carbaldehyde with aminopropylated silica followed by stirring of the polymer with a solution of either CuCl or CuBr furnished dark brown catalysts 72 (Scheme 31) which were active in ATRC of trichloro-, dichloro- and monobromo-substrates.33 Both efficient 5-exo and 5-endo cyclisations could be mediated using this reagent system. However, the supported catalyst was less active than its equivalent homogeneous counterpart 52a. Hence, reaction of the solid-supported catalyst with N-tosyl-N-allyl-2,2,2-trichloroacetamide 12b in dichloromethane at room temperature furnished the expected atom transfer product 13b in 92% after 3 hours. The catalyst
was reclaimed by filtration and was then re-used with a new batch of 12b to give 13b in 90% yield after 18–24 hours. The decrease in activity of the catalyst system upon re-use was not found to be due to leaching of the copper from the polymer bound catalysts but due to the formation of an inactive CuCl2 complex. A wide range of substrates could be cyclised including secondary monohalo-substrates and unsubstituted amides (Scheme 32). The diastereoselectivity of the processes was similar to that observed in homogenous catalysis.33
 |
| | Scheme 31 | |
 |
| | Scheme 32 | |
7 Perfluorous substituted ATRC catalysts
The use of perfluorous catalysis in a variety of organic reactions has been developed. Generally two phases (a perfluorous solvent and an organic solvent) are used in the reactions. In this approach the perfluorinated catalyst is confined to the perfluorous phase while the organic reactants and products are confined to the organic phase. At elevated temperatures the two phases are miscible allowing the desired reaction to be catalysed while at low (normally ambient) temperatures the phases are immiscible thus allowing easy separation of products from catalysts. In addition the inertness of most perfluorous solvents makes them relatively environmentally friendly. The perfluorinated analogue 74 of Me6-tren 49 was shown to be soluble in perfluorocyclohexane.34 The ligand itself was used as a 1 ∶ 1 mixture with CuCl and screened in the macrocyclisation reaction of trichloroester 38a. Cyclisation was much slower using
74 in the perfluorous solvent than when the corresponding non-perfluorinated ligand in 1,2-dichloroethane was used under convenional ATRC conditions. However, reaction of 38a with 5 mol% of the catalysts derived from CuCl and 74 furnished 39a in 97% yield after 10 hours (Scheme 33).34 Recycling of the catalysts was accomplished by simple decantation of the perfluorous layer under argon. Even after the fourth recycling protocol the catalyst was still active and led to no drop in ultimate conversion (run 1, 96%; run 2, 95%; run 3, 93%; run 4, 91%), although the reaction rate was retarded slightly. Analysis by atomic absorption spectroscopy indicated that only 1–2% of the copper was leached into the organic phase during each run. The addition of iron powder to the reactions was shown to improve the yield of the reactions.34
 |
| | Scheme 33 | |
 |
| | Scheme 34 | |
 |
| | Scheme 35 | |
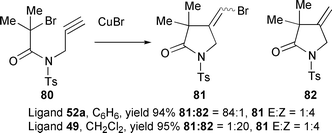
Cyclisation of the mono-bromo precursor 80 gave both the expected atom transfer product 81 plus the reduced product 82 arising from abstraction of a hydrogen atom by the intermediate vinyl radical (Scheme 36). The ratio of these compounds was dependent upon both the solvent and/or ligand employed for the reaction. Hence, cyclisation with CuCl–52a in benzene gave a 84 ∶ 1 ratio of 81
∶
82 in 94% combined yield while reaction with CuBr–49 in THF furnished the reduced compound 82 as the major product 81
∶
82 = 1 ∶ 20.21,25
 |
| | Scheme 36 | |
9 5-endo Cyclisations. Radical–polar crossover reactions
While 5-exo cyclisations have been extensively studied, the mediation of the less common 5-endo cyclisations has attracted less interest. Recently Zard has employed his Ni metal procedure to mediate cyclisation of trichloroacetamides in a 5-endo mode with the products being similar to those obtained using copper catalysis.35 Treatment of trichloro-substrate 83 with either CuCl–bipy36,37 or CuCl–52a26 has been reported to give the diene 84 (in 94 and 70% yields, respectively) (Scheme 37). Monohalo-substrate 85 furnished two alkene products 86 and 87 in 82% yield (1 ∶ 1 ratio) (Scheme 38).26 Mechanistically the reaction has been postulated to take place via an initial 5-endo radical
cyclisation to give 88 followed by Cu(II) mediated oxidation of the tertiary radical to the corresponding N-acyliminium ion 89 followed by elimination of a proton to furnish the two regioisomers 86 and 87.26
 |
| | Scheme 37 | |
 |
| | Scheme 38 | |
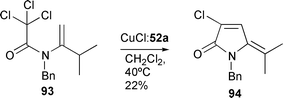
The reaction was found to be general with a variety of ring sizes and ring substitutions (Scheme 39).26,36,37 Reaction of the enamide 90 at 110 °C furnished the diene 92 arising through cyclisation of the thermodynamically more stable alternative enamide 91 (Scheme 40).36 Presumably the isomerisation to the thermodynamic enamide 91 is more rapid than cyclisation at this temperature. In contrast, cyclisation of trichloro-substrate 93 at lower temperature underwent exclusive cyclisation via the kinetic enamide (Scheme 41).26
 |
| | Scheme 39 | |
 |
| | Scheme 40 | |
 |
| | Scheme 41 | |
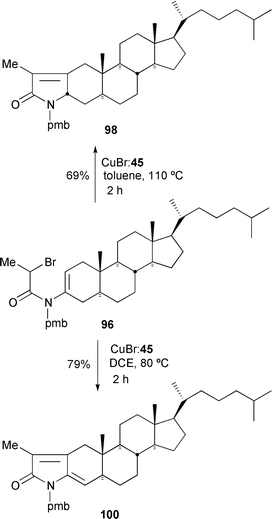
While it was not possible to mediate 5-endo cyclisation of secondary monohalo-substrates (e.g. 95) using ligands 49 and 52a,26 it was possible using the tripyridyl ligand 45.38 Reaction of enamides 95 and 96 with 1 equivalent of CuBr–45 in refluxing toluene gave the α,β-unsaturated lactams 97 and 98 in 86 and 69%, respectively (Schemes 42 and 43). Interestingly, if 1,2-dichloroethane was used instead, the corresponding dienes 99 and 100 were producted exclusively.38
 |
| | Scheme 42 | |
 |
| | Scheme 43 | |
These dienes are likely to be produced by further oxidation of the monoenes 97 and 98 under these reaction conditions as demonstrated by the transformation of 101 to 102 upon heating with 1 equivalent CuBr–45 (Scheme 44).38 Attempts to trap out the postulated acyliminium ion with nucleophiles have met with limited success. Hence, reaction of 93 with CuCl–52a in the presence of MeOH furnished the lactam 103, albeit in only low yield (30%) (Scheme 45).
 |
| | Scheme 44 | |
 |
| | Scheme 45 | |
10 Synthesis of β-lactams
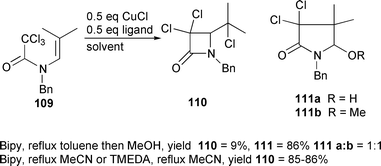
Enamides substituted only at the terminal end of the alkene group undergo rapid ATRC to furnish β-lactams in high yield.39,40 Cyclisation of 104 using 30 mol% CuBr–45 in CH2Cl2 at room temperature furnished the desired atom transfer product 105 in 94% yield after only 1 hour.39 Facile elimination of the tertiary bromide to furnish the alkene 106 was possible by reaction with DBU (Scheme 46). Cyclisation of secondary monobromide 107 required heating for 24 hours. Under these conditions the alkene 108 was obtained directly as a 2.1 ∶ 1 ratio of diastereomers (Scheme 47). Presumably initial atom transfer is followed by rapid elimination at the elevated temperature of the reaction. The regiochemical outcome of cyclisation had been reported to be solvent and
ligand dependent.40 Thus heating 109 with 0.5 equivalents of CuCl–bipy in toluene led to only 9% of the β-lactam 110 and 86% of the corresponding γ-lactams 111 (Scheme 48). However, when the reaction was repeated using CuCl–bipy or CuCl–TMEDA in acetonitrile as solvent only the β-lactam was produced. The change in regioselectivity can be explained by the differences in the solubilities of the copper catalysts. Thus because the CuCl–bipy catalyst is relatively insoluble in toluene, rapid trapping of the initial kinetically produced 4-exo cyclised radical is inefficient and this radical has time to undergo ring opening and thermodynamic 5-endo ring closure. However in acetonitrile (where the catalyst system is much more soluble) the initial kinetically produced radical undergoes highly efficient trapping leading to the β-lactam only.
 |
| | Scheme 46 | |
 |
| | Scheme 47 | |
 |
| | Scheme 48 | |
4-exo Cyclisation has also been observed in systems where the cyclised radical is particularly stabilised.26 Hence, 4-exo cyclisation of 112 gives the benzylic radical 113 which upon atom transfer furnished the bromofunctionalised β-lactam 114 in 99% yield as a 1 ∶ 1 mixture of diastereomers (Scheme 49).
 |
| | Scheme 49 | |
11 Concluding remarks
The last decade has seen the emergence of copper mediated atom transfer radical cyclisation reactions as a useful tool in the construction of cyclic molecules. Cyclisation of a range of substrates is possible furnishing a versatile array of ring sizes (4–18 membered rings), often in high yields. The development of activated complexes that mediate the cyclisation of monohalo substrates at room temperature or below, coupled with ability to sequence both intra- and intermolecular additions has broadened the scope of this technology. In particular, the development of solid supported/polymer bound reagents as well perfluorinated complexes makes the new processes relatively attractive towards industrial applications. Future investigations into the use of dendritic reagents, and the application of ATRC in ‘environmentally friendly’ solvents (such as water or ionic liquids) will further increase the attractiveness of this technology. The recent development of radical–polar
crossover reactions (particularly in 5-endo processes) is particularly valuable. The potential to sequence both radical reactions and cationic reactions increases the number of possible synthetic applications of this methodology.
References
- W. R. Bowman, C. F. Bridge and P. Brookes, J. Chem. Soc., Perkin Trans. 1, 2000, 1 RSC.
- S. Murai, N. Sonoda and S. Tsutsumi, J. Org. Chem., 1964, 29, 2104 CAS.
- J. Boivin, M. Yousfi and S. Z. Zard, Tetrahedron Lett., 1994, 35, 5629 CrossRef CAS.
- J. Iqbal, B. Bhatia and N. K. Nayyar, Chem. Rev., 1994, 94, 519 CrossRef CAS.
- H. Nagashima, H. Wakamatsu, K. Itoh, Y. Tomo and J. Tsuji, Tetrahedron Lett., 1983, 24, 2395 CrossRef CAS.
- N H. Nagashima, H. Wakamatsu and K. Itoh, J. Chem. Soc., Chem. Commun., 1984, 652 RSC.
- H. Nagashima, K. Ara, H. Wakamatsu and K. Itoh, J. Chem. Soc., Chem. Commun., 1985, 518 RSC.
- H. Nagashim, K. Seji, N. Ozaki, H. Wakamatsu, K. Itoh, Y. Tomo and J. Tsuji, J. Org. Chem., 1990, 55, 986.
- H. Nagashim, N. Ozaki, M. Ishii, K. Seki, M. Washiyama and K. Itoh, J. Org. Chem., 1993, 58, 464 CrossRef.
- S. Iwamatsu, K. Matsubara and H. Nagashima, J. Org. Chem., 1999, 64, 9625 CrossRef CAS.
- H. Nagashima, Y. Isono and S. Iwamatsu, J. Org. Chem., 2001, 66, 315 CrossRef CAS.
- J. H. Udding, C. J. M. Tuijp, H. Hiemstra and W. N. Speckamp, Tetrahedron, 1994, 50, 1907 CrossRef CAS.
- J. H. Udding, K. J. M. Tuijp, M. N. A. van Zanden, H. Hiemstra and W. N. Speckamp, J. Org. Chem., 1994, 59, 1993 CrossRef CAS.
- R. N. Ram and I. Charles, Chem. Commun., 1999, 2267 RSC.
- N. Baldovini, M.-P. Bertand, A. Carriere, R. Nouguier and J.-M. Plancher, J. Org. Chem., 1996, 61, 3205 CrossRef CAS.
- F. O. H. Pirrung, W. J. M. Steeman, H. Hiemstra, W. N. Speckamp, B. Kaptein, W. H. J. Boesten, H. E. Schoemaker and J. Kamphius, Tetrahedron Lett., 1992, 33, 5141 CrossRef CAS.
- F. O. H. Pirrung, H. Hiemstra, B. Kaptein, M. L. M. Sobrino, D. G. I. Petra, H. E. Schoemaker and W. N. Speckamp, Synlett, 1993, 50, 735.
- F. O. H. Pirrung, H. Hiemstra, W. N. Speckamp, B. Kaptein and H. E. Schoemaker, Tetrahedron, 1994, 50, 12415 CrossRef CAS.
- F. De Campo, D. Lastécouères and J.-B. Verlhac, Chem. Commun., 1998, 2117 RSC.
- F. De Campo, D. Lastécouères and J.-B. Verlhac, J. Chem. Soc., Perkin Trans. 1, 2000, 575 RSC.
- A. J. Clark, F. De Campo, R. J. Deeth, R. P. Filik, S. Gatard, N. A. Hunt, D. Lastécouères, G. H. Thomas, J.-B. Verlhac and H. Wongtap, J. Chem. Soc., Perkin Trans. 1, 2000, 671 RSC.
- D. M. Haddleton, A. J. Clark, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, J. Chem. Soc., Dalton Trans., 1998, 381 RSC.
- A. J. Clark, D. J. Duncalf, R. P. Filik, D. H. Haddleton, G. H. Thomas and H. Wongtap, Tetrahedron Lett., 1999, 40, 3807 CrossRef CAS.
- A. J. Clark, G. M. Battle, A. M. Heming, D. M. Haddleton and A. Bridge, Tetrahedron Lett., 2001, 42, 2003 CrossRef CAS.
- A. J. Clark, G. M. Battle and A. Bridge, Tetrahedron Lett., 2001, 42, 1999 CrossRef CAS.
- A. J. Clark, C. P. Dell, J. M. Ellard, N. A. Hunt and J. P. McDonagh, Tetrahedron Lett., 1999, 40, 8619 CrossRef CAS.
- M. Benedetti, L. Forti, F. Ghelfi, U. M. Pagnoni and R. Ronzoni, Tetrahedron., 1997, 41, 14031 CrossRef CAS.
- F. Ghelfi, F. Bellesia, L. Forti, G. Ghirardini, R. Grandi, E. Libertini, M. C. Montemaggi, U. M. Pagnoni, A. Pinetti, L. De Buyck and A. F. Parsons., Tetrahedron, 1999, 55, 5839 CrossRef CAS.
- F. Ghelfi and A. F. Parsons, J. Org. Chem., 2000, 65, 6249 CrossRef CAS.
- F. Ghelfi, G. Ghirardini, E. Libertini, L. Forti and U. M. Pagnoni, Tetrahedron Lett., 1999, 40, 8595 CrossRef CAS.
- A. J. Clark, R. P. Filik and G. H. Thomas, Tetrahedron Lett., 1999, 40, 4885 CrossRef CAS.
- S. Iwamatsu, H. Kondo, K. Matsubara and H. Nagashima, Tetrahedron, 1999, 55, 1687 CrossRef CAS.
- A. J. Clark, R. P. Filik, D. M. Haddleton, A. Radigue, C. J. Sanders, G. H. Thomas and M. E. Smith, J. Org. Chem., 1999, 64, 8954 CrossRef CAS.
- F. De Campo, D. Lastécouères, J.-M. Vincent and J.-B. Verlhac, J. Org. Chem., 1999, 64, 4969 CrossRef CAS.
- J. Cassayre, B. Quiclet-Sire, J.-B. Saunier and S. Z. Zard, Tetrahedron, 1998, 54, 1029 CrossRef CAS.
- D. T. Davies, N. Kapur and A. F. Parsons, Tetrahedron Lett., 1999, 40, 8615 CrossRef CAS.
- D. T. Davies, N. Kapur and A. F. Parsons, Tetrahedron, 2000, 56, 3941 CrossRef CAS.
- A. J. Clark, C. P. Dell and J. P. McDonagh, C. R. Acad. Sci. Ser IIc: Chim., 2001, 4, 575 CrossRef CAS.
- A. J. Clark, G. M. Battle and A. Bridge, Tetrahedron Lett., 2001, 42, 4409 CrossRef CAS.
- J. S. Bryans, N. E. A. Chessum, A. F. Parsons and F. Ghelfi, Tetrahedron Lett., 2001, 42, 2901 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here.