Celastraceae sesquiterpenoids: biological activity and synthesis
Alan C. Spivey, Matthew Weston and Steven Woodhead
Department of Chemistry, Brook Hill, Sheffield, UK S3 7HF. E-mail: a.c.spivey@sheffield.ac.uk;; Fax: (0114) 2738673;; Tel: (0114) 2229467
First published on 13th December 2001
Abstract
Plant extracts of the Celastraceae have been used for centuries throughout South America and China as insect repellents and insecticides in traditional agriculture, and also for the treatment of a plethora of medical ailments from stomach complaints and fever to rheumatoid arthritis and cancer. Many of the medicinally interesting properties associated with these crude preparations have now been attributed to a large family of highly oxygenated sesquiterpenoids based on a tricyclic dihydroagarofuran skeleton. In this article, the structural diversity and range of biological activities associated with this intriguing class of natural products are examined with a view to stimulating interest in their total synthesis. Existing synthetic endeavours towards their synthesis are also evaluated.
 Alan C. Spivey Alan C. Spivey | Alan Spivey (left) received a BSc in chemistry in 1988 from the University of Nottingham and a DPhil in 1991 from the University of Oxford under the guidance of Professor Sir Jack Baldwin. After post-doctoral work at the Université de Genève, Switzerland with the late Professor Wolfgang Oppolzer, and at the University of Cambridge with Professor Sir Alan Battersby, he moved to a lectureship at the University of Sheffield where he is currently a reader. Research in his group is focused on the development of new catalysts for asymmetric acylation, new linkers for solid phase organic synthesis, chemical aspects of signal transduction, and total synthesis, e.g. of biologically active Celastraceae sesquiterpenoids. |
Matthew Weston (centre) received an MChem in chemistry in 1998 from the University of Sheffield and is currently in the third year of research towards a PhD exploring approaches to the total synthesis of bioactive Celastraceae sesquiterpenoid cores under the direction of Alan Spivey. |
Steven Woodhead (right) received a BSc in chemistry in 1995 and his PhD in 1999, under the direction of Alan Spivey, at the University of Sheffield where he initiated the Celastraceae total synthesis program. After post-doctoral research at Florida State University with Professor Robert Holton, he returned to the UK for a further period of post-doctoral study with Professor Gerald Pattenden at the University of Nottingham. He is currently working in medicinal chemistry research with Astex-technology in Cambridge, UK. |
1 Background and introduction to the Celastraceae
The Celastraceae family is indigenous to tropical and sub-tropical regions of the world, including North Africa, South America, and many parts of East Asia, particularly China.1,2 The family constitutes approximately 88 genera and 1300 species of plants.3 These plants generally grow as small trees, bushes, or lianas and have resinous stems and leaves. They have been valued since antiquity because their extracts have useful medicinal properties.4 Indeed, the variety of properties attributed to crude plant extracts of the Celastraceae in traditional medicine and agriculture is astonishing, and includes stimulant, appetite suppressive, sedative, emetic, purgative, memory-restorative, male contraceptive, anti-tumour, anti-leukemic, anti-bacterial, insecticidal and insect repellent activities!5 For example, extracts of the bark of the thunder-god vine Tripterygium wilfordii have been used by the Chinese for centuries to combat life-threatening illness, and have more recently found application in the treatment of leukemia and rheumatoid arthritis.6 Similarly, the stimulating properties of the leaves of the khat bush Catha edulis, which grows in certain parts of East Africa and Southern Arabia, were reported in the literature as early as 1237 by the physician Naguib Ad Din, who proposed the use of khat for alleviating depression.7 Since khat leaves rapidly lose their effect after picking, the culture of chewing khat leaves for their stimulant/euphoric and appetite/fatigue suppressive effects was for centuries confined to the immediate areas where the plant grows.7 With improvements in road communications, however, the habit had become so widespread by the mid-1970s that a form of addiction known as khatism was beginning to present serious social and economic difficulties in Ethiopia and the Yemen Republic.4 This prompted the World Health Organisation to fund an investigation to determine the active constituents of khat. It was this program,8 and the co-incidental discovery in 1972 of the macrolide maytansine from Maytenus ovatus, which displayed exceptionally high inhibitory activity in vitro against certain human carcinomas,9 that catapulted the natural product chemistry of the Celastraceae into the limelight.Over the last 30 years or so, a large number of secondary metabolites exhibiting a wide range of bioactivity have been extracted from the Celastraceae. In addition to numerous terpenoids, including a diverse array of sesquiterpenoids which constitute the focus of this article, various bioactive phenylalkylamines, maytansinoids, and flavonoids have also been isolated5,10 (Fig. 1).
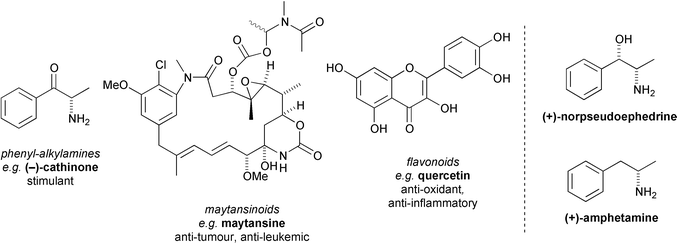 | ||
| Fig. 1 Non-terpenoid secondary metabolites of the Celastraceae, and the structures of norpseudoephedrine and amphetamine. | ||
In particular, the α-aminoketone (−)-cathinone has now been established to be the unstable constituent of fresh khat leaves which is primarily responsible for their stimulant and euphoric effects.10 (−)-Cathinone is structurally related to, and has the same absolute configuration as, (+)-amphetamine and (+)-norpseudoephedrine but is a more potent stimulant.7 The development of maytansine† and related macrolides as anti-tumour leads was halted in the 1980s when they were found to cause serious gastro-intestinal damage in rats,5 but recent advances in drug targeting via conjugation to cancer-specific humanised antibodies appear to offer new opportunities for clinical application.11 The flavonoid quercetin, which is also found in a number of other plant families, displays a wide spectrum of anti-oxidant and anti-inflammatory activity as a result of its ability to quench reactive oxygen radicals.10
The bulk of the bioactive constituents of the Celastraceae, however, are terpenoids. Terpenoids are a structurally diverse class of secondary metabolites composed from an integral number of biological equivalents of the C5 hydrocarbon isoprene (2-methylbuta-1,3-diene, Fig. 2). During their biosynthesis these ‘monomers’ are joined together ‘head-to-tail’ fashion, frequently with extensive subsequent modification of structure due to rearrangements and functional group changes to give characteristic and topologically related families of natural products.12 Thus, monoterpenoids comprise two isoprene units and therefore have a C10 core skeleton, sesquiterpenoids three isoprene units (C15), diterpenoids four isoprene units (C20), and triterpenoids six isoprene units (C30). All these types of terpenoids are found in extracts of the Celastraceae (Fig. 2).
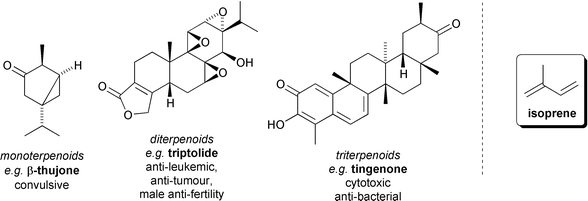 | ||
| Fig. 2 Mono-, di-, and triterpenoid metabolites of the Celastraceae, and the structure of isoprene. | ||
Monoterpenoids such as β-thujone are volatile compounds responsible for the characteristic odour of e.g. freshly picked khat leaves.10 The tri-epoxide diterpenoid triptolide was isolated from the roots of the thunder-god vine Tripterygium wilfordii, and has been shown to be responsible for some of the significant anti-leukemic and anti-tumour activity of this plant.13 Triptolide also displays interesting male anti-fertility properties and is one of a number of diterpene epoxides contained in a commercially available ‘total multi-glycoside extract’ of Tripterygium wilfordii that has been used clinically for male fertility control in China.14,15 The triterpene tingenone and related quinone methides display cytotoxic and anti-bacterial activity which is believed to be the result of DNA intercalation and nucleophilic addition by DNA bases.16
The most pervasive and characteristic metabolites of the Celastraceae, however, are a large family of unusually highly oxygenated sesquiterpenoids, and it is this class of compounds that has attracted the most interest. The remainder of this article overviews the structural diversity displayed by this class of natural products, the extent of their biological activities, and details the most significant synthetic endeavours towards their total synthesis. Particular attention is focused on methods used to construct the tetrahydrofuran C-ring of the dihydroagarofuran skeleton (see next section), as this structural feature is not found in other sesquiterpenoids.
2 The diversity of Celastraceae sesquiterpenoid structures
Celastraceae sesquiterpenoids generally occur as poly-esters of variously poly-oxygenated tricyclic scaffolds all based on a core C15 skeleton known as dihydroagarofuran.17–19‡ This skeleton comprises A and B rings in the form of an axially dimethylated trans-decalin bicycle with a 1,3-diaxially fused, Me2C–O bridge constituting the tetrahydrofuranyl C-ring. Throughout this review the dihydroagarofuran ring system is drawn such that the C-ring is on the bottom face of the molecule, as depicted unboxed on the left-hand side of Fig. 3. This elevation has been used because it corresponds to that most commonly employed in literature of the Celastraceae, and particularly in the seminal isolation studies of Crombie et al.,4 the defining synthetic studies of White et al.,20 and the extensive biological screening studies of Takaishi et al.6 (vide infra).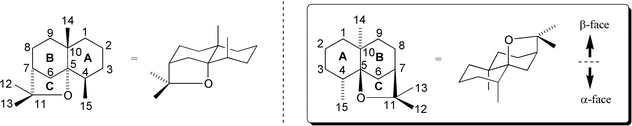 | ||
| Fig. 3 The dihydroagarofuran skeleton. | ||
It is important to note, however, that the alternative elevation in which the C-ring is on the top face of the molecule, as depicted in the box on the right-hand side of Fig. 3, corresponds to the conventional presentation of terpenoid and steroid structures.§ A number of authors including Brüning,1 Büchi,18 Gonzalez,21 and Huffman22 have adhered to this convention. Two numbering regimes have also been employed in the literature of the Celastraceae. The most prevalent by far, and that used throughout this review, is illustrated in Fig. 3.¶ The alternative regime is defined in the publications of Yamada23 and Takaishi.6 The enantiomer of agarofuran depicted in Fig. 3 has the correct absolute configuration for naturally occurring (−)-dihydroagarofuran,17 and all the dihydroagarofurans of the Celastraceae for which determinations have been made [e.g. (−)-euonyminol24–27].
Well over 200 members of this chemical class have now been characterised, based on over 40, variously oxygenated dihydroagarofuran core structures. These core structures can have as few as two additional oxygen containing groups, as in the case of boariol, or as many as nine, as in the case of euonyminol (Fig. 4).2,4,5
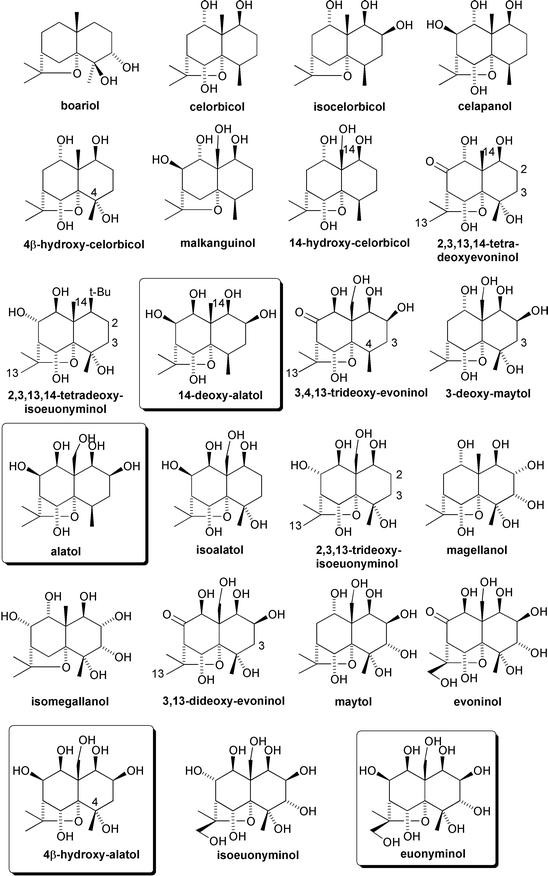 | ||
| Fig. 4 Some polyhydroxy dihydroagarofuran sesquiterpenoid cores. Those with symmetric hydroxylation across the ‘northern’ periphery are boxed. | ||
The peripheral esterifying residues vary widely from acetic (Ac) and benzoic (Bz) acids through more complex aliphatic and heterocyclic acids to stereochemically elaborate pyridine-containing dicarboxylic acids (and a monoterpene diacid)15 which invariably form macrodilactone bridges between C-3 and C-13 and/or C-8 and C-14 (Fig. 5). The relative position, number, and configuration of these residues distinguishes each individual sesquiterpenoid.||
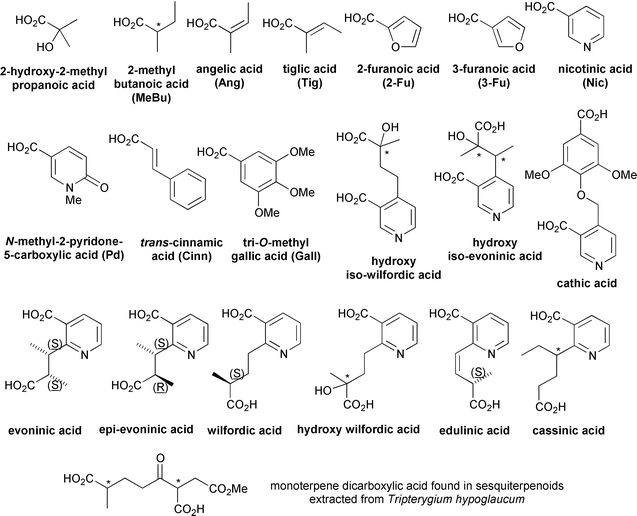 | ||
| Fig. 5 Esterifying residues found in Celastraceae sesquiterpenoids. | ||
Four core structures: 14-deoxyalatol, alatol, 4β-hydroxyalatol, and euonyminol have been highlighted in Fig. 4. This is because they have a symmetric array of hydroxylation across the α-face of their ‘northern’ periphery. As will become apparent in the following overview of biological activity, Celastraceae natural products comprising these cores invariably display interesting activity. It was this observation, combined with the notion that such latent symmetry might be exploited to facilitate their synthesis, which inspired our own synthetic program towards the total asymmetric synthesis of these Celastraceae sesquiterpenoid cores (see Scheme 13).
3 Biological activity of the Celastraceae sesquiterpenoids28
3.1 Insecticidal and insect anti-feedant activity
Various species of Celastraceae have been used in traditional Chinese agriculture for centuries to protect crops against insect damage.29 The first insecticidal constituents were isolated from extracts of Tripterygium wilfordii and characterised as the sesquiterpenoids wilfordine and wilforine.30 These alkaloids, which are based on the euonyminol core, were found to be highly toxic towards the larvae of the European corn borer Pyrausta nubilalis at concentrations of 60 ppm (100% mortality after 3 days).31 Additionally, wilforine displays effective anti-feedant activity towards cabbage leaf worm Pieris rapae at 1 × 10−5% dry weight, and migratory locusts Locusta migratoria at 5 × 10−5% dry weight.32 This makes wilforine more potent towards these pests than azadirachtin, the most notable insect anti-feedant from plants (which is not a Celastraceae natural product, but which comes from the Indian Neem tree Azadirachta indica)32 (Fig. 6).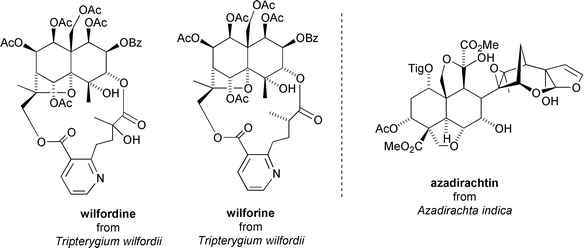 | ||
| Fig. 6 Insecticidal/anti-feedant Celastraceae sesquiterpenoids from Tripterygium wilfordii and the structure of azadirachtin. | ||
The powdered root bark of Chinese bittersweet Celastrus angulatus has also yielded a number of anti-feedant sesquiterpenes, most notably celangulin which is based on the 4β-hydroxyalatol core.33 Celangulin exhibits activity against the fall armyworm Mythimna separata at 5 ppm and against the cabbage leaf worm Pieris rapae at 222 ppm.33 The seed oil of Euonymus bungeanus and the powdered seeds of Euonymus fortunei have also yielded two alatol-based sesquiterpenoids which display activity against Pieris rapae at 200 and 1000 ppm, respectively.34 A series of macrodilactone-containing sesquiterpenoids, known as cathedulins and which are based on the euonyminol core, have been isolated from the leaves and twigs of the khat bush Catha edulis.4 Many of the cathedulins possess significant insect anti-feedant properties. In particular, cathedulin E-5** exhibits potent activity against the pink bollworm Pectinophora gossypiella at 1 ppm, a value comparable with that of azadirachtin35 (Fig. 7).
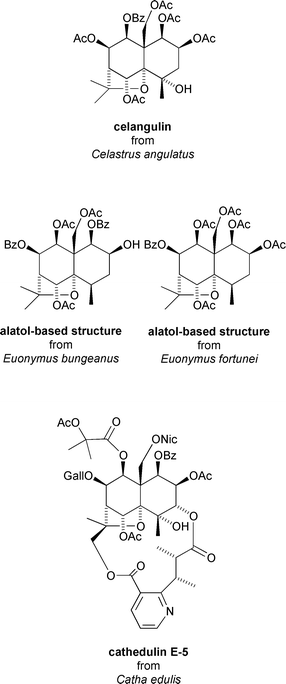 | ||
| Fig. 7 Insecticidal/anti-feedant sesquiterpenoids from Celastrus angulatus, Euonymus bungeanus, Euonymus fortunei and Catha edulis. | ||
Recently, Gonzalez carried out a large and systematic study of insecticidal and anti-feedant activity of sesquiterpenes sourced from a variety of species of Celastraceae using the leaf disk method against the Egyptian cotton leaf worm Spodoptera littoralis.29 Most of the agarofuran sesquiterpenes tested showed activity at 1 μg cm−2, but the most active ones, based on the 4β-hydroxyalatol and isoalatol cores, and including celangulin, were found to be active at 0.1 μg cm−2, and in one case at 0.01 μg cm−2, which is more potent than the well-known anti-feedant triphenyl tin acetate29 (Fig. 8).
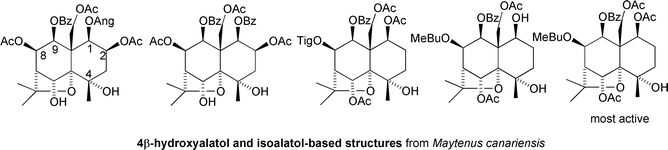 | ||
| Fig. 8 Anti-feedant sesquiterpenoids from Maytenus canariensis. | ||
For compounds with the same skeleton, activity was found to vary with the nature of the esterifying residues.29 The greater the number of acetyl residues, the greater the activity. The opposite was observed with benzoyl residues, where increasing frequency was observed to decrease anti-feedant activity. Compounds with aromatic esters at C-1 or C-9 were found to be only weakly active, while the introduction of esterifying residues at C-2 had no observable effect upon activity. Insecticidal activity was roughly correlated with the presence of a carbonyl group at C-8, with sesquiterpenoids having this feature isolated from Maytenus canariensis and Orthosphenia mexicana being the most potent.29 The evoninol skeleton appeared to confer the greatest activity (LD100 = 0.01 μg cm−2)††29 (Fig. 9).
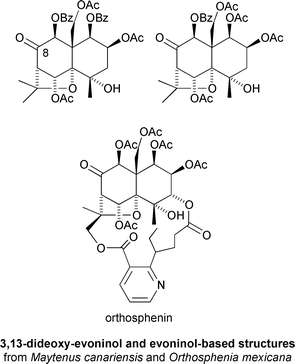 | ||
| Fig. 9 Insecticidal sesquiterpenoids from Maytenus canariensis and Orthosphenia mexicana. | ||
3.2 Cytotoxicity and anti-cancer activity
Plants of the genus Tripterygium wilfordii have been used in traditional Chinese medicine for centuries for the treatment of cancer.21 The presence of the anti-tumour and anti-leukemic diterpenoid triptolide13 (Fig. 2), and the cytotoxic triterpene tingenone16 (Fig. 2) in this genus accounts for much of this activity. However, the discovery of the anti-tumour properties of the macrolide maytansine from Maytenus ovatus9 (Fig. 1), promoted a widespread search for further Celastraceae-derived anti-cancer agents. The first to be found was celaglaumin from the bark of Celastrus glaucophyllus which was found to exhibit weak activity against the murine leukemia cell line L1210 (IC50 = 2.11 μg mL−1)‡‡ and the lymphatic leukemia cell line P-388 (IC50 = 4.12 μg mL−1).36 More recently, Takaishi has evaluated a number of dihydroagarofuran sesquiterpenes using an Epstein-Barr virus early antigen (EBV-EA) screen and found that triptofordin F-2 and triptogelin A-1 from Tripterygium wilfordii show promising anti-tumour promoting activity.37 Triptogelin A-1 was shown to reduce both the incidence and frequency of skin tumours in mice without excessive cytotoxicity. From these studies it was deduced that the functionalities at C-6, C-14 and C-8 play an important part in the inhibition of EBV: an acetyl residue at C-6 confers greater activity compared to an alcohol, while a nicotinoyl residue is optimal. The presence of C-14 as an acetoxymethyl group also appears to confer superior activity compared to having a methyl group at this position37 (Fig. 10).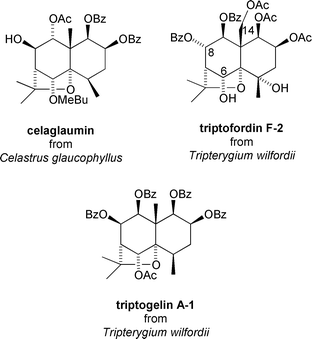 | ||
| Fig. 10 Anti-tumour promoting sesquiterpenoids from Celastrus glaucophyllus and Tripterygium wilfordii. | ||
EBV-EA activation inhibition has also been reported for celafolins D-2 and B-2 isolated from the seeds of Celastrus stephanotiifolius,38 and for a series of 2α,4β-dihydrocelorbicol and 3-deoxymaytol-based sesquiterpenoids from the fruits of Maytenus cuzcoina21 (Fig. 11).
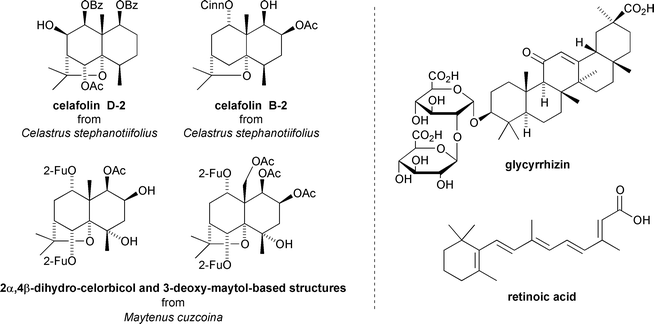 | ||
| Fig. 11 Anti-tumour promoting sesquiterpenoids from Celastrus stephanotiifolius and Maytenus cuzcoina, and the structures of the anti-tumour promoters glycyrrhizin and retinoic acid. | ||
The Maytenus cuzcoina-derived compounds show more significant inhibition in Raji cells than standard anti-tumour promoters such as glycyrrhizin and retinoic acid.21 The 3-deoxymaytol-based derivative was found to be the most active and moreover, inhibitory activity within the dihydrocelorbicol series was strongly influenced by the nature of esterification at the C-2 position: OAc > O(2-Fu) > OH > OMeBu.21
A series of dilactone bridged dihydroagarofuran structures, including emarginatine-A and emarginatinine, which were isolated from the stems and branches of Maytenus emarginata, have also been shown to display in vitro cytotoxicity against the human epidermoid carcinoma of the nasopharynx KB (ED50 = 0.4–4.0 μg mL−1).§§39 Emarginatine-A, which is based on the euonyminol skeleton and contains a C-3–C-13 evononic acid dilactone bridge, was slightly less active than emarginatinine, which differs only by containing a C-3–C-13 hydroxywilfordic acid dilactone bridge (ED50 = 4.0 vs. 2.1 μg mL−1, respectively).39 Emarginatine-F, which is based on the isoeuonyminol skeleton, showed more potent activity against the human epidermoid carcinoma of the nasopharynx KB (ED50 = 0.51 μg mL−1), and also showed significant activity against the human colon adenocarcinoma cell line HCT-8 (ED50 = 1.29 μg mL−1), the human medulloblastoma cell line TE-671 (ED50 = 0.21 μg mL−1), and the murine lymphocytic leukemia cell line P-388 (ED50 = 0.69 μg mL−1).40 A related macrolide, celahinine A, was also found to be present in a crude ethanolic extract of Celastrus hindsii which displayed potent in vitro cytotoxicity against human nasopharynx-, cervix-, and colon-carcinoma cell lines41 (Fig. 12).
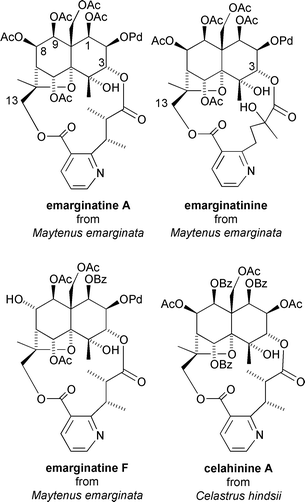 | ||
| Fig. 12 Cytotoxic sesquiterpenoids from Maytenus emarginata and Celastrus hindsii. | ||
3.3 Reversal of multi-drug resistance (MDR)
Celastraceae sesquiterpenoids have also been found to display interesting reversal of multi-drug resistance (MDR) in cancer cells. MDR is a major problem in chemotherapy treatments for various cancers.42 A number of compounds, including celafolin A-1 and celorbicol, have been isolated from Celastrus orbiculatus which reverse, either partially or totally, resistance to adriamycin, vinblastine, and paclitaxel in the human nasopharynx carcinoma cells KB-V1 and the human breast cancer cells MCF7/ADR.43 These sesquiterpenoids were found to be less toxic and of greater reversing power than other known reversing agents such as verapamil that are currently approved for clinical use42 (Fig. 13).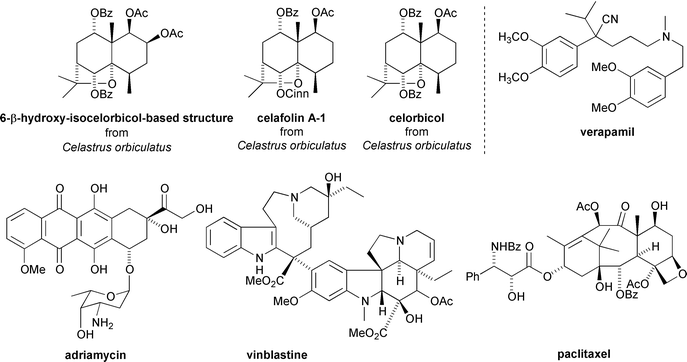 | ||
| Fig. 13 MDR reversing sesquiterpenoids from Celastrus orbiculatus and the structures of MDR reversing agent verapamil and cancer chemotherapy agents adriamycin, vinblastine and paclitaxel. | ||
More recently, a series of sesquiterpenoids based on 4β,14-dihydroxycelapanol and 3,13-dideoxyeuonyminol cores have been isolated from the aerial parts of Crossopetalum tonduzzi and Maytenus macrocarpa.44 These have been found to reverse the resistance of parasitic protazoan Leishmania tropica to growth inhibition by daunomycin (75–95% at 15 μM concentration with respect to uninhibited control growth)44 (Fig. 14).
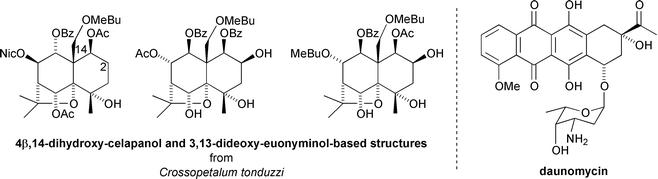 | ||
| Fig. 14 MDR reversing sesquiterpenoids from Crossopetalum tonduzzi and the structure of cancer chemotherapy agent daunomycin. | ||
The size of the substituent at C-2 was suggested to be important for MDR activity.44 The most active compounds were those with a proton or alcohol substituent at C-2: esterification at this site resulted in reduced activity. In contrast, a benzoyl at C-1 imparted greater activity than an acetyl.44
3.4 Immunosuppressive activity
Immunosuppressive agents are key components within the cocktail of drugs administered during organ transplant surgery.45 Promising immunosuppressive activity has been reported for two sesquiterpenoid constituents, wilfortrine and euonine,46 and a triterpene constituent, celastrol,45 of Tripterygium wilfordii. The sesquiterpenoids are based on the euonyminol core, have C-3–C-13 hydroxywilfordic acid dilactone bridges, and have been found to elicit marked depressant effects on humoral-mediated immunity using the hemolysin reaction index on mice (wilfortrine, 40 mg kg−1; euonine, 80 mg kg−1).46 Both sesquiterpenoids significantly decreased charcoal particle clearance and the weights of spleen and thymus in the mice relative to healthy control animals at 80 mg kg−146 (Fig. 15).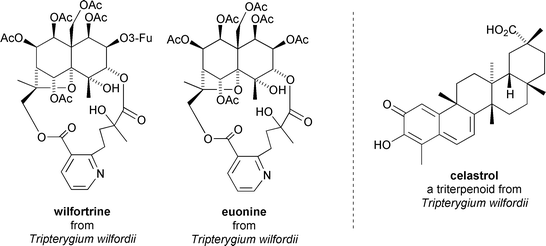 | ||
| Fig. 15 Immunosuppressive sesquiterpenoids from Tripterygium wilfordii and the structure of immunosuppressive triterpenoid celastrol. | ||
3.5 Anti-HIV activity
Perhaps the most exciting biological activity associated with the Celastraceae sesquiterpenoids is that of selective anti-HIV activity. Takaishi has recently reported that various macrodilactone containing dihydroagarofuran derivatives, and in particular, hypoglaunine B, triptonine B, and hyponine B extracted from the root bark of Tripterygium hypoglaucum, exhibit potent inhibition of HIV virus replication in human lymphocyte cells.15 All three sesquiterpenoids, and also wilfortrine (Fig. 16), display EC50 values¶¶ of <0.01 μg mL−1 and in vitro therapeutic indexes (TIs)|||| >1000.15 In general a TI >5.0 is considered significant. By way of comparison clinically approved anti-HIV agent AZT has an EC50 value of <0.012 μg mL−1 and TI of ∼42000 under these conditions.15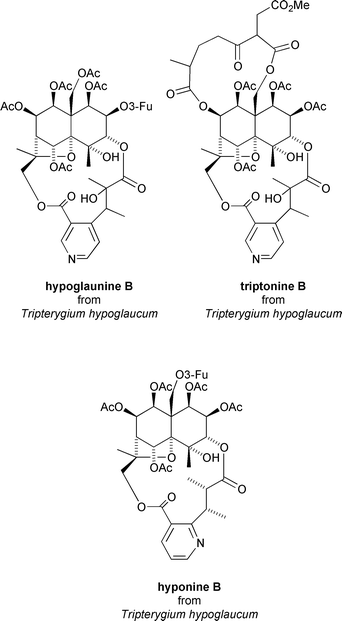 | ||
| Fig. 16 Anti-HIV sesquiterpenoids from Tripterygium hypoglaucum. | ||
It is apparent from the foregoing, by no means exhaustive,28 overview of biological activities associated with polyester dihydroagarofuran extracts of the Celastraceae that these intriguing compounds possess a rich and diverse spectrum of medicinally interesting activity. However, low natural abundance coupled with isolation difficulties continues to impede exhaustive biological screening and the establishment of meaningful structure–activity relationships for this class of natural products. Consequently, their molecular modes of action remain unknown. It is unlikely that this situation will be rectified unless robust and flexible asymmetric syntheses of the core structures are developed. This would pave the way for the total synthesis of selected fully esterified natural products and hopefully allow the identification of pharmacophore leads for drug development. However, synthetic endeavours in this area to date have been relatively scarce (see next section). This probably reflects the fact that many of these compounds have complex structures containing dense contiguous functionality and the lack of a single ‘high-profile’ target structure (cf. e.g. taxol) to attract synthetic chemists. Significant biological activity appears to encompass a plethora of stereochemical permutations around the dihydroagarofuran core.28 However, it is striking that many of the most biologically active members of this class have a symmetric array of hydroxylation across the α-face of their northern periphery. It is hoped that this observation will inspire further efforts towards the synthesis of these intriguing compounds.
4 Synthetic approaches to Celastraceae sesquiterpenoids47
4.1 Early synthetic work—syntheses of the agarofuran skeleton
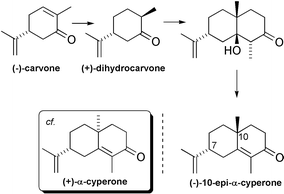
The first agarofuran synthesis reported was that of (+)-α-agarofuran by Barrett and Büchi in 1967.48 Their synthesis employed readily available (−)-10-epi-α-cyperone as starting material.*** The overall efficiency of their synthesis was, however, compromised by the formation of a mixture of triene isomers (1 and 2) early in the synthesis (only one of which underwent photooxygenation), and by a final dehydration step which afforded the undesired alkene isomer 5 as a major by-product. Of particular note in their synthesis was the facile alumina-catalysed cyclisation reaction of the axial C-5 tertiary alcohol of 3 onto C-11 of the axial isopropenyl double bond in 4 used to close the C-11–O bond of the tetrahydrofuran C-ring (Scheme 1).![The Büchi synthesis of (+)-α-agarofuran (1967). Reagents and yields: (i) LiAlH4 [81%]; (ii) Al2O3–Pyr [81%, total]; (iii) O2, hv [62%]; (iv) basic Al2O3 [84%]; (v) acidic Al2O3 [72%]; (vi) NaBH4 [88%]; (vii) SOCl2; (viii) LiAlH4 [87%, total].](/image/article/2002/CS/b000678p/b000678p-s1.gif) | ||
| Scheme 1 The Büchi synthesis of (+)-α-agarofuran (1967). Reagents and yields: (i) LiAlH4 [81%]; (ii) Al2O3–Pyr [81%, total]; (iii) O2, hv [62%]; (iv) basic Al2O3 [84%]; (v) acidic Al2O3 [72%]; (vi) NaBH4 [88%]; (vii) SOCl2; (viii) LiAlH4 [87%, total]. | ||
Subsequently, Marshall and Pike reported a more efficient synthesis of (+)-α-agarofuran from (−)-10-epi-α-cyperone in a sequence of six steps.50 In contrast to Barrett and Büchi these workers installed the tetrahydrofuran C-ring by C-5–O bond formation. Thus, cyclisation of the C-11 isopropene-derived tertiary alcohol onto the C-4–C-5 alkene (6 → 7) was triggered by treatment with MCPBA, presumably via ring opening of an intermediate α-epoxide (which could not be detected). They also attempted to develop a method for the conversion of (+)-α-agarofuran to (−)-β-agarofuran. However, they found that irradiation of their product using a high-pressure Hg vapour lamp effected only partial photochemical isomerisation to a 3∶7 mixture of α- and β-agarofurans50 (Scheme 2).
![The Marshall synthesis of (+)-α-agarofuran (1968). Reagents and yields: (i) MCPBA; (ii) LiAlH4; (iii) Ac2O; (iv) Li, NH3 [83%, 4 steps]; (v) MCPBA. [55%]; (vi) SOCl2 [94%]; (vii) hv, xylene–IPA [100%, total].](/image/article/2002/CS/b000678p/b000678p-s2.gif) | ||
| Scheme 2 The Marshall synthesis of (+)-α-agarofuran (1968). Reagents and yields: (i) MCPBA; (ii) LiAlH4; (iii) Ac2O; (iv) Li, NH3 [83%, 4 steps]; (v) MCPBA. [55%]; (vi) SOCl2 [94%]; (vii) hv, xylene–IPA [100%, total]. | ||
A shorter synthesis of (+)-α-agarofuran was reported by Deslongchamps et al.51 using the dehydration precursor of (−)-10-epi-α-cyperone as starting material.*** In this synthesis, as in Marshall and Pike’s, the tetrahydrofuran C-ring was installed by C-5–O bond formation [9 → (+)-α-agarofuran]. In this case, however, the C-4–C-5 alkene formed part of an allylic alcohol system in 9, and so cyclisation of the isopropene-derived C-11 tertiary alcohol could be effected by acid-catalysed intramolecular substitution with allylic rearrangement. This ring-closure was high yielding, but the efficiency was compromised by the co-formation of an uncyclised diene. An analogous acid-catalysed ring-closure was also shown to be possible when the C-4–C-5 alkene formed part of an enone system (8 → 10). Although this ring-closure reaction was considerably lower yielding it enabled the first synthesis of (−)-isodihydroagarofuran51 (Scheme 3).
![The Deslongchamps synthesis of (+)-α-agarofuran (1968). Reagents and yields: (i) Hg(OAc)2, NaOH, NaBH4 [75%]; (ii) NaOMe [81%]; (iii) LiAl(OtBu)3H [100%]; (iv) PTSA [100%, total]; (v) PTSA [30%]; (vi) toluene-p-sulfonylhydrazine, then NaBH4 [25%].](/image/article/2002/CS/b000678p/b000678p-s3.gif) | ||
| Scheme 3 The Deslongchamps synthesis of (+)-α-agarofuran (1968). Reagents and yields: (i) Hg(OAc)2, NaOH, NaBH4 [75%]; (ii) NaOMe [81%]; (iii) LiAl(OtBu)3H [100%]; (iv) PTSA [100%, total]; (v) PTSA [30%]; (vi) toluene-p-sulfonylhydrazine, then NaBH4 [25%]. | ||
In 1972 Kelly reported a diastereoselective route to (±)-nor-ketoagarofuran, which is an analogue of β-agarofuran having a C-4 ketone group in place of the C-4–C-15 alkene.52 The approach was an evolution of earlier endeavours by Heathcock and Kelly in which the final steps had turned out to be capricious and irreproducible.53 Kelly employed an interesting epimerisation–lactonization strategy to install the key tetrahydrofuran C-ring, and as such this was the first synthesis of an agarofuran which did not rely on (−)-10-epi-α-cyperone for provision of the crucial axial stereochemistry at C-7 of the B–C-ring junction. The early stages of the synthesis featured an unusual conversion of the equatorial C-7 alcohol into the corresponding chloride using PCl5 with retention of configuration as the result of neighbouring-group participation by the C-4–C-5 alkene (11 → 12). Once this chloride was converted to the corresponding methyl ester, inversion of this centre was achieved by the ingenious base-mediated epimerisation step driven by lactonisation onto the axial C-5 tertiary alcohol and removal of methanol (13 → 14). Conversion of lactone 14 into the corresponding dimethyl tetrahydrofuran C-ring involved treatment with MeLi followed by acid-catalysed ring-closure of the resulting C-11–C-5 diol [15 → (±)-nor-ketoagarofuran]52 (Scheme 4).
![The Kelly synthesis of (±)-nor-ketoagarofuran (1972). Reagents and yields: (i) Na, MeCOCH2CH2NEt2 [35%]; (ii) BzCl; (iii) NaBH4 [60%, 2 steps]; (iv) PCl5 [72%]; (v) Mg then CO2 [78%]; (vi) HCO2H–H2O2; (vii) NaOH [50%, 2 steps]; (viii) Jones’ reagent [93%]; (ix) PTSA, HO(CH2)2OH [42%]; (x) LiOMe–BzOEt, MeOH [70%]; (xi) LiOMe–BzOEt, dioxane (xii) MeLi; (xiii) HCl [65%, 3 steps].](/image/article/2002/CS/b000678p/b000678p-s4.gif) | ||
| Scheme 4 The Kelly synthesis of (±)-nor-ketoagarofuran (1972). Reagents and yields: (i) Na, MeCOCH2CH2NEt2 [35%]; (ii) BzCl; (iii) NaBH4 [60%, 2 steps]; (iv) PCl5 [72%]; (v) Mg then CO2 [78%]; (vi) HCO2H–H2O2; (vii) NaOH [50%, 2 steps]; (viii) Jones’ reagent [93%]; (ix) PTSA, HO(CH2)2OH [42%]; (x) LiOMe–BzOEt, MeOH [70%]; (xi) LiOMe–BzOEt, dioxane (xii) MeLi; (xiii) HCl [65%, 3 steps]. | ||
The first stereocontrolled synthesis of (−)-β-agarofuran was reported by Büchi and Wüest in 1979.18 Their synthesis employed (−)-carvone as starting material and was unusual in that the tetrahydrofuran C-ring was installed relatively early in the sequence. Thus, C-11–O bond formation was achieved by intramolecular 1,2-addition of the isopropene-derived C-11 tertiary alcohol to a C-5 ketone upon treatment with PCl5 (16 → 17). This set the stage for the penultimate step, an elegant radical cyclisation of the A-ring with concomitant installation of a C-4–C-15 vinylsilane as a mixture of stereoisomers (17 → 18). This inseparable mixture of vinylsilanes was readily converted into the requisite exomethylene function of (−)-β-agarofuran on treatment with acid [18 → (−)-β-agarofuran]. A further important discovery by these workers was that (−)-β-agarofuran could be hydrogenated with >95∶5 diastereoselectivity to (−)-dihydroagarofuran using diimide (cf. standard catalytic hydrogenation which is much less selective). Pre-coordination of the diimide to the tetrahydrofuran ether oxygen was implicated as being responsible for the high selectivity18 (Scheme 5).
![The Büchi synthesis of (−)-β-agarofuran (1979). Reagents and yields: (i) aq. H2SO4 [63%]; (ii) 10% Pd/C–H2 [93%]; (iii) KNH2 [41%]; (iv) PCl5 [71%]; (v) EtMgBr then TMSCl [98%]; (vi) AIBN, HSnBu3, hv [72%, total]; (vii) PTSA [92%]; (viii) hydrazine, H2O2 [92%].](/image/article/2002/CS/b000678p/b000678p-s5.gif) | ||
| Scheme 5 The Büchi synthesis of (−)-β-agarofuran (1979). Reagents and yields: (i) aq. H2SO4 [63%]; (ii) 10% Pd/C–H2 [93%]; (iii) KNH2 [41%]; (iv) PCl5 [71%]; (v) EtMgBr then TMSCl [98%]; (vi) AIBN, HSnBu3, hv [72%, total]; (vii) PTSA [92%]; (viii) hydrazine, H2O2 [92%]. | ||
4.2 Synthetic approaches to polyoxygenated agarofurans
Following these pioneering efforts by the groups of Büchi, Marshall, Deslongchamps, and Kelly, the late 1970s and early 1980s saw Huffmann et al. take up the challenge of developing these foundations into an approach to the more highly oxygenated agarofuran derivatives found in the Celastraceae. After a number of twists and turns, this work culminated in the total synthesis of (±)-isocelorbicol in 1987.22 Huffmann’s final approach to (±)-isocelorbicol began with a three-step synthesis of (±)-10-epi-9-oxo-α-cyperone from (−)-carvone via (±)-hydroxycarvone.††† This sequence featured a Robinson annulation reaction analogous to the one used for the formation of (−)-10-epi-α-cyperone from (+)-dihydrocarvone.*** (±)-10-Epi-α-cyperone was converted into 9-keto-α-agarofuran by a method closely similar to that used in the 9-deoxy series by Deslongchamps et al., but employing Jones’ reagent to effect the C-11–O bond formation for tetrahydrofuran C-ring formation (19 → 20). The remainder of the synthesis proceeded with high levels of stereocontrol and featured a diimide reduction of the C-4–C-15 alkene in the manner of Büchi’s β-agarofuran synthesis (21 → 22) and a totally diastereoselective dihydroxylation of the C-1–C-2 alkene on the α-face using stoichiometric OsO4 (23 → 24). The overall yield of (±)-isocelorbicol over the fifteen steps from 9-keto-α-agarofuran was 3.2% (Scheme 6).![The Huffmann synthesis of (±)-isocelorbicol (1986). Reagents and yields: (i) KOH–H2O2 then HCl [60%]; (ii) EVK, KOH; (iii) PTSA [32%, 2 steps]; (iv) MCPBA; (v) LiAlH4; (vi) Jones’ reagent [47%, 3 steps]; (vii) MCPBA, NaHCO3 [70%]; (viii) LDA [72%]; (ix) NH2NH2, NaIO4, CuSO4, AcOH [90%]; (x) POCl3 [65%]; (xi) LiAlH4; (xii) BuLi then BzCl [93%, 2 steps]; (xiii) MCPBA [60%]; (xiv) (PhSe)2, NaBH4 [44%]; (xv) MCPBA, iPr2NH [87%]; (xvi) OsO4, Pyr [66%]; (xvii) 2,2-DMP, CSA [87%]; (xviii) NaH, imidazole then CS2 then MeI; (xix) nBu3SnH [82%, 2 steps]; (xx) Ba(OH)2; (xxi) HCl [86%, 2 steps].](/image/article/2002/CS/b000678p/b000678p-s6.gif) | ||
| Scheme 6 The Huffmann synthesis of (±)-isocelorbicol (1986). Reagents and yields: (i) KOH–H2O2 then HCl [60%]; (ii) EVK, KOH; (iii) PTSA [32%, 2 steps]; (iv) MCPBA; (v) LiAlH4; (vi) Jones’ reagent [47%, 3 steps]; (vii) MCPBA, NaHCO3 [70%]; (viii) LDA [72%]; (ix) NH2NH2, NaIO4, CuSO4, AcOH [90%]; (x) POCl3 [65%]; (xi) LiAlH4; (xii) BuLi then BzCl [93%, 2 steps]; (xiii) MCPBA [60%]; (xiv) (PhSe)2, NaBH4 [44%]; (xv) MCPBA, iPr2NH [87%]; (xvi) OsO4, Pyr [66%]; (xvii) 2,2-DMP, CSA [87%]; (xviii) NaH, imidazole then CS2 then MeI; (xix) nBu3SnH [82%, 2 steps]; (xx) Ba(OH)2; (xxi) HCl [86%, 2 steps]. | ||
No significant further developments were made towards the synthesis of more highly oxygenated dihydroagarofurans until the early 1990s. At this time two Chinese groups began to report a series of detailed studies towards the synthesis of functionalised agarofurans. The groups of Li54 and Guo55 have now developed a number of modifications and extensions to the syntheses described above, from (−)-carvone and (±)-hydroxycarvone, so as to access oxygenated agarofuran derivatives containing up to three additional oxygen functions (Scheme 7).
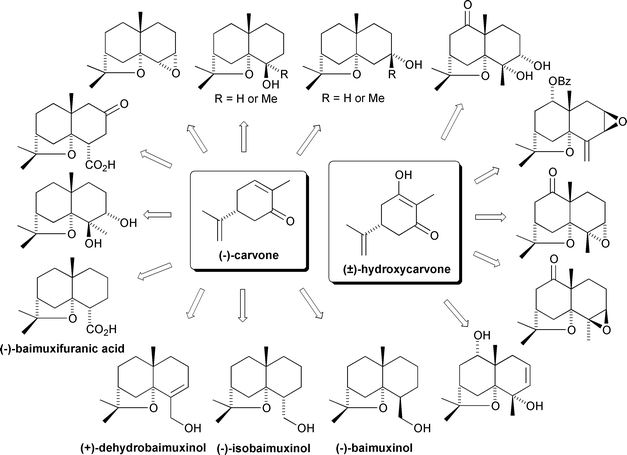 | ||
| Scheme 7 Overview of the Li and Guo syntheses of oxygenated agarofurans (1990s). | ||
Of particular note from this corpus of work was Li’s optimisation of Huffmann’s (+)-α-agarofuran and (±)-9-keto-α-agarofuran syntheses by rendering the Robinson annulation reactions more diastereoselective and improving the C-11–O bond formation for tetrahydrofuran C-ring cyclisation by the use of H2SO4 in place of Jones’ reagent.49 Additionally, Li has employed the Shapiro reaction‡‡‡ to convert the tosyl hydrazone of 3-keto-4β,9β-dihydroxydihydroagarofuran 25 into the corresponding 2,3-unsaturated compound 26 in an approach towards 1α,9β-dibenzoyloxy-2β,3β,4β-trihydroxydihydroagarofuran 27.56–58 This target compound is a natural product from Salvia palaefoliais which is used in Colombia as an anti-hypertensive agent58 (Scheme 8).
![The Li synthesis of (±)-2,3-dehydro-4β,9β-dihydroxydihydroagarofuran (1995). Reagents and yields: (i) OsO4, Pyr [99%]; (ii) DMSO, TFAA, Et3N [75%]; (iii) TsNHNH2 [100%]; (iv) LDA [70%].](/image/article/2002/CS/b000678p/b000678p-s8.gif) | ||
| Scheme 8 The Li synthesis of (±)-2,3-dehydro-4β,9β-dihydroxydihydroagarofuran (1995). Reagents and yields: (i) OsO4, Pyr [99%]; (ii) DMSO, TFAA, Et3N [75%]; (iii) TsNHNH2 [100%]; (iv) LDA [70%]. | ||
More recently, the group of Tu59 has explored the use of the readily available sesquiterpene (−)-α-santonin as a conveniently functionalised starting material for the synthesis of oxygenated dihydroagarofurans.§§§ Of particular interest from these studies is that, in contrast to all the approaches mentioned above, the use of this starting material allows differentiation of the C-12 and C-13 substituents of the tetrahydrofuran C-ring. This is required for the synthesis of the most highly oxygenated Celastraceae target cores (e.g. euonyminol and evoninol, see Fig. 4) which have C-12 as a methyl group and C-13 as a hydroxymethyl group. The orchestration of synthetic steps that allowed for this differentiation started with an unusual rearrangement of the C-5–C-6 epoxy lactone 28 to the C-7–C-11 alkenyl lactone 29 on treatment with NaOMe. This transformation collapses the C-7 stereocentre (which held the C-7–C-11 lactone bond in the undesirable equatorial orientation in 28) to give the tetrasubstituted C-7–C-11 alkene in 29 with complete stereoselectivity. At a later stage, a rather neat cyclisation of the axial C-5 tertiary alcohol onto the Si-face of this alkene at C-11 is employed to close the C-11–O bond of the agarofuranyl tetrahydrofuran C-ring. This cyclisation sets up the correct axial configuration at C-7 and establishes C-12 and C-13 as methyl and hydroxymethyl groups, respectively. Initial studies used a mercuric acetate-mediated ring-closure reaction followed by reductive demercuration for this purpose (30 → 31),61 but subsequently a cleaner NBS-mediated ring-closure protocol, followed by nBu3SnH-mediated dehalogenation was disclosed (32 → 33).62 This method was used for the preparation of their most advanced intermediate to date: the 3-keto-6,8,9,13-tetrahydroxyagarofuran derivative 3459 (Scheme 9).
![The Tu approach to dihydroagarofurans from (−)-α-santonin (1998, 1999). Reagents and yields: (i) H2, Raney-Ni [80%]; (ii) PTSA, HO(CH2)2OH [66%]; (iii) MCPBA [68%]; (iv) NaOMe [83%]; (v) LiAlH4 [95%]; (vi) Hg(OAc)2; (vii) NaBH4 [82%, 2 steps]; (viii) PTSA, acetone; (ix) LiAlH4 [70%, 2 steps]; (x) PhCHO, ZnCl2 [72%]; (xi) SeO2, tBuOOH; [82%]; (xii) NBS, CaCO3 [95%]; (xiii) nBu3SnH [92%]; (xiv) SOCl2, Pyr [52%]; (xv) NMO, OsO4 [87%].](/image/article/2002/CS/b000678p/b000678p-s9.gif) | ||
| Scheme 9 The Tu approach to dihydroagarofurans from (−)-α-santonin (1998, 1999). Reagents and yields: (i) H2, Raney-Ni [80%]; (ii) PTSA, HO(CH2)2OH [66%]; (iii) MCPBA [68%]; (iv) NaOMe [83%]; (v) LiAlH4 [95%]; (vi) Hg(OAc)2; (vii) NaBH4 [82%, 2 steps]; (viii) PTSA, acetone; (ix) LiAlH4 [70%, 2 steps]; (x) PhCHO, ZnCl2 [72%]; (xi) SeO2, tBuOOH; [82%]; (xii) NBS, CaCO3 [95%]; (xiii) nBu3SnH [92%]; (xiv) SOCl2, Pyr [52%]; (xv) NMO, OsO4 [87%]. | ||
Notwithstanding the important contributions made by the aforementioned groups, the most significant synthesis in the Celastraceae sesquiterpenoid area has undoubtedly been White et al.’s synthesis of (±)-euonyminol in 1997.20 This synthesis opened with a Diels–Alder reaction between an in situ prepared benzoquinone and a siloxypentadiene which established the basic decalin A–B ring system 35. The crucial axial stereochemistry at C-7 of the B–C-ring junction was established by an unusual conjugate addition reaction of iPrMgBr to the β-face of the B-ring enone directed by coordination to the C-6 hydroxy group (36 → 37). Subsequent epoxidation of the isopropenyl double bond (37 → 38),¶¶¶ again directed by the proximal C-6 hydroxy group, set the stage for an impressive acid-mediated cascade reaction. This cascade reaction (38 → 39), which formed the cornerstone of White’s synthesis, not only closed the tetrahydrofuran C-ring by C-11–O bond formation, it also established C-12 and C-13 as methyl and hydroxymethyl, respectively, and installed oxygenation at C-2 with the correct α-orientation. Elaboration of the cascade product 39 into the highly oxygenated (±)-euonyminol Celastraceae core was achieved, generally with high levels of stereocontrol, by a further series of elegant steps. Particular highlights included an aldol–retro-aldol reaction sequence to invert the C-1 hydroxy substituent (40 → 41) and the use of a Me3Al-mediated Lobry de Bruyn–Alberda van Eckenstein rearrangement to accomplish 1, 2-transposition of the C-8–C-9 vicinal hydroxyketone (42 → 43). Although initial attempts to effect the final cis-dihydroxylation of the C-3–C-4 alkene lacked stereocontrol, a change of protecting group regime to that shown in Scheme 10 (i.e.44 → 45) resulted in almost exclusive β-face attack as required.20
![The White synthesis of (±)-euonyminol (1997). Reagents and yields: (i) Ag2O [94%]; (ii) NBS, Bz2O2; (iii) Et3N [98%, 2 steps]; (iv) NaBH4, CeCl3·7H2O [90%]; (v) MCPBA [88%]; (vi) LDA, 15-crown-5 [63%]; (vii) tBuOOH, 2,6-lutidine, VO(acac)2 [74%]; (viii) TFA; (ix) Pyr–THF–H2O then imidazole, MeCN [75%, 2 steps]; (x) PPTS, PhCH(OMe)2 [83%]; (xi) nBu4NF [96%]; (xii) TBSOTf, Et3N [100%]; (xiii) KHMDS [86%]; (xiv) AlMe3 [100%]; (xv) LiAlH4 then HCl; (xvi) TBSOTf, Et3N; (xvii) OsO4, Pyr; (xviii) Amberlite IR-120, AcOH–H2O; (xix) Ac2O, Pyr, [30%, 5 steps]; (xx) NaOMe, MeOH, then Amberlite IR-120 [100%].](/image/article/2002/CS/b000678p/b000678p-s10.gif) | ||
| Scheme 10 The White synthesis of (±)-euonyminol (1997). Reagents and yields: (i) Ag2O [94%]; (ii) NBS, Bz2O2; (iii) Et3N [98%, 2 steps]; (iv) NaBH4, CeCl3·7H2O [90%]; (v) MCPBA [88%]; (vi) LDA, 15-crown-5 [63%]; (vii) tBuOOH, 2,6-lutidine, VO(acac)2 [74%]; (viii) TFA; (ix) Pyr–THF–H2O then imidazole, MeCN [75%, 2 steps]; (x) PPTS, PhCH(OMe)2 [83%]; (xi) nBu4NF [96%]; (xii) TBSOTf, Et3N [100%]; (xiii) KHMDS [86%]; (xiv) AlMe3 [100%]; (xv) LiAlH4 then HCl; (xvi) TBSOTf, Et3N; (xvii) OsO4, Pyr; (xviii) Amberlite IR-120, AcOH–H2O; (xix) Ac2O, Pyr, [30%, 5 steps]; (xx) NaOMe, MeOH, then Amberlite IR-120 [100%]. | ||
Very recently Ducrot et al. have described the synthesis of various racemic functionalised decalins designed to be advanced intermediates for the preparation of polyhydroxylated Celastraceae sesquiterpenoid cores.63 The synthesis of the most elaborate intermediate 49 uses the cyclohexane-1,4-dione derivative 46 as starting material. This is converted to a conveniently functionalised decalin enone 48 in a series of six steps featuring a neat oxidative cleavage of a furan ring with concomitant intra-molecular aldol condensation (47 → 48) (Scheme 11).
![The Ducrot synthesis of a functionalised (±)-decalin derivative (1999). Reagents and yields: (i) NaH, CO(OMe)2; (ii) PhSeCl, Pyr; (iii) H2O2 [66%, 3 steps]; (iv) furan, BF3 [61%]; (v) DMDO; (vi) TBSOTf, 2,6-lutidine [72%, 2 steps]; (vii) H2O2, NaOH [77%]; (viii) LiAlH4 [56%]; (ix) PTSA [40%]; (x) TBSOTf, 2,6-lutidine; (xi) LiBH4; (xii) TBSOTf, 2,6-lutidine; (xiii) TFA [94%, 4 steps]; (xiv) MeLi [36%].](/image/article/2002/CS/b000678p/b000678p-s11.gif) | ||
| Scheme 11 The Ducrot synthesis of a functionalised (±)-decalin derivative (1999). Reagents and yields: (i) NaH, CO(OMe)2; (ii) PhSeCl, Pyr; (iii) H2O2 [66%, 3 steps]; (iv) furan, BF3 [61%]; (v) DMDO; (vi) TBSOTf, 2,6-lutidine [72%, 2 steps]; (vii) H2O2, NaOH [77%]; (viii) LiAlH4 [56%]; (ix) PTSA [40%]; (x) TBSOTf, 2,6-lutidine; (xi) LiBH4; (xii) TBSOTf, 2,6-lutidine; (xiii) TFA [94%, 4 steps]; (xiv) MeLi [36%]. | ||
Ducrot et al. have also disclosed some model studies for tetrahydrofuran C-ring closure of potential utility later in their synthesis.64 Essentially two strategies were explored for conversion of the B-ring enone functionality of readily available (±)-Wieland–Miescher ketone into the B–C-ring junction of an agarofuran. The first method proceeded via the trans-decalin vinyl nitrile 50 and afforded the simple ring-closed agarofuran 55, whereas the second method proceeded via the cis-decalin vinyl nitrile 51 and also introduced a β-hydroxy group at C-6 to give agarofuran 57, albeit in substantially lower yield.64 Both methods bore similarities to the Kelly approach to tetrahydrofuran C-ring-closure (cf.Scheme 4). Thus, the first method exploited a base-mediated α- to β-face epimerisation of a C-7 aldehyde substituent driven by lactol formation (52 → 53), followed by aqueous H2SO4-mediated ring-closure of the C-11–C-5 diol 54 to form the tetrahydrofuran C-ring in agarofuran 55. In the second method analogous ring-closure of the C-11–C-5 diol 56 to form the tetrahydrofuran C-ring in agarofuran 57 was orchestrated to be accompanied by ring-opening of the C-6–C-7 α-epoxide by water thereby concomitantly introducing the C-6 β-hydroxy group (along with an unwanted tertiary α-hydroxy group at C-7)64 (Scheme 12).
![The Ducrot strategy for tetrahydrofuran ring-closure (2000). Reagents and yields: (i) NaBH4 [98%]; (ii) TBSCl, imidazole [92%]; (iii) DIBAL-H [97%]; (iv) (NO2)2C6H3CO2H, DEAD, PPh3; (v) K2CO3 [75%, 2 steps]; (vi) MCPBA [51%, 2 steps]; (vii) TsCl, Pyr [82%]; (viii) NaCN, HMPA [79%]; (ix) 10% Pd/C, H2 [80%]; (x) DIBAL-H [42%, total]; (xi) Al2O3 (basic) [100%]; (xii) Ag2CO3, Celite [72%]; (xiii) MeLi [68%]; (xiv) H2SO4, acetone [65%]; (xv) MeLi; (xvi) PPTS [75%, 2 steps]; (xvii) MeLi [80%]; (xviii) MCPBA [91%]; (xix) H2SO4 [39%].](/image/article/2002/CS/b000678p/b000678p-s12.gif) | ||
| Scheme 12 The Ducrot strategy for tetrahydrofuran ring-closure (2000). Reagents and yields: (i) NaBH4 [98%]; (ii) TBSCl, imidazole [92%]; (iii) DIBAL-H [97%]; (iv) (NO2)2C6H3CO2H, DEAD, PPh3; (v) K2CO3 [75%, 2 steps]; (vi) MCPBA [51%, 2 steps]; (vii) TsCl, Pyr [82%]; (viii) NaCN, HMPA [79%]; (ix) 10% Pd/C, H2 [80%]; (x) DIBAL-H [42%, total]; (xi) Al2O3 (basic) [100%]; (xii) Ag2CO3, Celite [72%]; (xiii) MeLi [68%]; (xiv) H2SO4, acetone [65%]; (xv) MeLi; (xvi) PPTS [75%, 2 steps]; (xvii) MeLi [80%]; (xviii) MCPBA [91%]; (xix) H2SO4 [39%]. | ||
Our own approach to the synthesis of Celastraceae cores having symmetric oxygenation patterns across the northern periphery of the α-face of the dihydroagarofuran ring system (e.g. 14-deoxyalatol, alatol, 4β-hydroxyalatol, and euonyminol, see Fig. 4) centres around a ‘late-stage’ enantioselective desymmetrisation of meso-decalin diallylic alcohol 59.65 It is the first approach yet described towards this class of natural product core by asymmetric synthesis. The starting epoxide 58 was readily prepared from naphthalene by Birch reduction then epoxidation. This epoxide 58 was then converted in a series of totally diastereoselective transformations in a two-directional fashion through to the key meso-diallylic alcohol 59. Enantioselective desymmetrisation of this diallylic alcohol was then effected using a novel Zr-modified Sharpless asymmetric epoxidation protocol which afforded the desymmetrised epoxy alcohol 60, containing eight contiguous chiral centres, in >95% ee65 (Scheme 13).
![The Spivey asymmetric synthesis of a functionalised decalin derivative (2001). Reagents and yields: (i) Et2AlCN [98%]; (ii) VO(acac)2, tBuOOH [87%]; (iii) BnBr, NaH, nBu4NI [83%]; (iv) Ph3PBr2 [87%]; (v) TBSOTf, 2,6-lutidine [98%]; (vi) DBU [98%]; (vii) OsO4, NMO [79%]; (viii) PTSA, DMP [95%]; (ix) K2OsO2(OH)4, K3Fe(CN)6, K2CO3, MeSO2NH2, quinuclidine [74%]; (x) PTSA, DMP [98%]; (xi) Na–NH3 then TBAF [91%]; (xii) MsCl, Et3N [99%]; (xiii) DBN [67%]; (xiv) Zr(OiPr)4·iPrOH, D-(−)-DIPT, tBuOOH [55%].](/image/article/2002/CS/b000678p/b000678p-s13.gif) | ||
| Scheme 13 The Spivey asymmetric synthesis of a functionalised decalin derivative (2001). Reagents and yields: (i) Et2AlCN [98%]; (ii) VO(acac)2, tBuOOH [87%]; (iii) BnBr, NaH, nBu4NI [83%]; (iv) Ph3PBr2 [87%]; (v) TBSOTf, 2,6-lutidine [98%]; (vi) DBU [98%]; (vii) OsO4, NMO [79%]; (viii) PTSA, DMP [95%]; (ix) K2OsO2(OH)4, K3Fe(CN)6, K2CO3, MeSO2NH2, quinuclidine [74%]; (x) PTSA, DMP [98%]; (xi) Na–NH3 then TBAF [91%]; (xii) MsCl, Et3N [99%]; (xiii) DBN [67%]; (xiv) Zr(OiPr)4·iPrOH, D-(−)-DIPT, tBuOOH [55%]. | ||
We are currently exploring novel strategies for elaboration of the C-6–C-7 alkene in decalin 60 to allow tetrahydrofuran C-ring formation with concomitant differentiation of C-12 and C-13, and introduction of a β-hydroxy group at C-6, as required for the preparation of (−)-euonyminol and (−)-evoninol.
5 Concluding remarks and outlook
It is hoped that the foregoing overview of synthetic work towards the synthesis of Celastraceae sesquiterpenoid natural products will provide a stimulus for further synthetic work in this area. Many challenges remain to be met, and in particular there remains the formidable task, over and above that of synthesis of the appropriate core structures, of devising appropriate strategies for the elaboration of naturally occurring esterification patterns around the periphery of these molecules. The architectural complexity presented, particularly by the macrodilactone-containing structures such as hypoglaunine B (Fig. 16), will undoubtedly require meticulous planning and judicious use of orthogonal protecting groups for success. To date there has been limited synthetic work carried out on the preparation of the functionalised nicotinate diacids (cf.Fig. 5)66 which constitute the backbone of many of these appendages1,4 and just one study relating to macrodilactone synthesis.23 This latter work was carried out by Yamada et al.23 who in 1975 performed a re-synthesis of (−)-evonine from a triol derivative of (−)-evoninol obtained from degradation studies. Stepwise formation of the 14-membered dilactone ring was accomplished by ester formation at the C-13 hydroxy group (61 → 62) followed by macrolactonisation onto the C-3 hydroxy group [63 → 64] (Scheme 14).![The Yamada re-synthesis of evonine (1975). Reagents and yields: (i) NaOMe [67%]; (ii) MeI, NaH [75%]; (iii) NaOMe [84%]; (iv) (MeO)2CMe2, CSA [56%]; (v) DMAP, Et3N, DME [34%]; (vi) 80% AcOH [82%]; (vii) CrO3, Pyr [70%]; (viii) AcOH [68%]; (ix) CH2N2; (x) NaH, DMF [12%, 2 steps]; (xi) BCl3; (xii) Ac2O, Pyr [35%, 2 steps].](/image/article/2002/CS/b000678p/b000678p-s14.gif) | ||
| Scheme 14 The Yamada re-synthesis of evonine (1975). Reagents and yields: (i) NaOMe [67%]; (ii) MeI, NaH [75%]; (iii) NaOMe [84%]; (iv) (MeO)2CMe2, CSA [56%]; (v) DMAP, Et3N, DME [34%]; (vi) 80% AcOH [82%]; (vii) CrO3, Pyr [70%]; (viii) AcOH [68%]; (ix) CH2N2; (x) NaH, DMF [12%, 2 steps]; (xi) BCl3; (xii) Ac2O, Pyr [35%, 2 steps]. | ||
Clearly there remains much challenging synthetic chemistry to be developed in order to provide compounds for biological screening. Impressive progress has, however, already been made and hopefully it will not be too long before synthetic material will be available to help unravel the biological modes of action of these molecules. Perhaps ultimately we may also be in a position to understand why nature defies entropy to create all these beautiful structures.
Note added in proof
Very recently Li et al. have published an asymmetric synthesis of (−)-isocelorbicol.67References
- R. Brüning and H. Wagner, Phytochemistry, 1978, 17, 1821, and references therein Search PubMed.
- O. Munoz, A. Penaloza, A. G. Gonzalez, A. G. Ravelo, I. L. Bazzocchi and N. L. Alvarenga, in Studies in Natural Products Chemistry, ed. Atta-ur-Rahman, Elsevier, 1996, vol. 18, pp. 739–783, and references therein Search PubMed.
- D. J. Mabberley, The Plant-book. A portable dictionary of the vascular plants, Cambridge University Press, Cambridge, 2nd edn., 1997 Search PubMed.
- L. Crombie, W. M. L. Crombie and D. A. Whiting, The Alkaloids, 1990, 39, 139, and references therein Search PubMed.
- L. Dubravkova, Z. Voticky and J. Tomko, Acta Fac. Pharm. Univ. Lomenianae, 1988, 42, 141, and references therein Search PubMed.
- H.-Q. Duan, Y. Takaishi, Y.-F. Jia and D. Li, Phytochemistry, 2001, 56, 341, and references therein Search PubMed.
- P. Kalix and O. Braenden, Pharmacol. Rev., 1985, 37, 149, and references therein Search PubMed.
- R. L. Baxter, L. Crombie, D. J. Simmonds, D. A. Whiting, O. J. Braenden and K. Szendrei, J. Chem. Soc., Perkin Trans. 1, 1979, 2965, and references therein Search PubMed.
- S. M. Kupchan, Y. Komoda, W. A. Court, G. J. Thomas, R. A. Smith, A. Karim, C. J. Gilmore and R. C. Haltiwanger, J. Am. Chem. Soc., 1972, 94, 1354 CrossRef CAS.
- K. Szendrei, Lausanne International Council on Alcohol and Addiction, 1983, 91, and references therein Search PubMed.
- C. Liu and R. V. J. Chari, Expert Opin. Invest. Drugs, 1997, 6, 169 Search PubMed.
- J. Mann, Secondary Metabolism, Clarendon Press, Oxford, 2nd edn., 1987 Search PubMed.
- D. Yang, X.-Y. Ye, M. Xu, K.-W. Pang, N. Zou and R. M. Letcher, J. Org. Chem., 1998, 63, 6446 CrossRef CAS.
- Q. S. Zhen, X. Ye and Z. J. Wei, Contraception, 1995, 51, 121 CrossRef CAS.
- H. Duan, Y. Takaishi, Y. Imakura, Y. Jia, D. Li, L. M. Cosentino and K.-H. Lee, J. Nat. Prod., 2000, 63, 357 CrossRef CAS.
- W. N. Setzer, M. T. Holland, C. A. Bozeman, G. F. Rozmus, M. C. Setzer, D. M. Moriarity, S. Reeb, B. Vogler, R. B. Bates and W. A. Haber, Planta Med., 2001, 67, 65 CrossRef CAS.
- M. L. Maheshwari, T. C. Jain, R. B. Bates and S. C. Bhattacharyya, Tetrahedron, 1963, 19, 1079, and references therein Search PubMed.
- G. Büchi and H. Wüest, J. Org. Chem., 1979, 44, 546, and references therein Search PubMed.
- M. L. Maheshwari, K. R. Varma and S. C. Bhattacharyya, Tetrahedron, 1963, 19, 1519 CrossRef CAS.
- J. D. White, H. Shin, T.-S. Kim and N. S. Cutshall, J. Am. Chem. Soc., 1997, 119, 2404 CrossRef CAS.
- A. G. Gonzalez, B. M. Tincusi, I. L. Bazzocchi, H. Tokuda, H. Nishino, T. Konoshima, I. A. Jimenez and A. G. Ravelo, Bioorg. Med. Chem., 2000, 8, 1773, and references therein Search PubMed.
- J. W. Huffman and P. C. Raveendranath, Tetrahedron, 1987, 43, 5565.
- K. Sugiura, K. Yamada and Y. Hirata, Chem. Lett., 1975, 6, 579.
- J.-H. Ryu, S. Y. Ryu, Y. N. Han and B. H. Han, Arch. Pharm. Res., 1997, 20, 76 Search PubMed.
- H. Itokawa, O. Shirota, H. Morita, K. Takeya and Y. Iitaka, J. Chem. Soc., Perkin Trans. 1, 1993, 1247 RSC.
- K. Sasaki and Y. Hirata, J. Chem. Soc., Perkin Trans. 2, 1972, 1268 RSC.
- M. Pailer and R. Libiseller, Monatsh., 1962, 93, 511 Search PubMed.
- For a recent review of biological activity of natural products of the Celastraceae see: A. G. Gonzalez, I. L. Bazzocchi, L. Moujir and I. A. Jimenez, Stud. Nat. Prod. Chem., 2000, 23, 649 Search PubMed.
- A. G. Gonzalez, I. A. Jiminez, A. G. Ravelo, J. Coll, J. A. Gonzalez and J. Lloria, Biochem. System. Ecol., 1997, 25, 513, and references therein Search PubMed.
- M. Beroza, J. Am. Chem. Soc., 1953, 75, 44, and references therein Search PubMed.
- M. Beroza and G. T. Bottger, J. Econom. Entomol., 1954, 47, 188, and references therein Search PubMed.
- F. Delle Monache, G. B. Marini Bettolo and E. A. Bernays, J. Appl. Entomol., 1984, 97, 406, and references therein Search PubMed.
- J.-K. Liu, Z.-J. Jia, D.-G. Wu, J. Zhou and Q.-G. Wang, Phytochemistry, 1990, 29, 2503 CrossRef CAS.
- Y.-Q. Tu, D.-G. Wu, J. Zhou, Y. Chen and S.-M. Hua, Acta Bot. Sin., 1991, 33, 876 Search PubMed; Y. Xiao and W. Xiajin, Nat. Prod. Res. Dev., 1994, 6, 37 Search PubMed.
- I. Kubo, M. Kim and G. De Boer, J. Chromatogr., 1987, 402, 354 CrossRef CAS.
- J. Zhou, J.-K. Liu, Z.-J. Jia, D.-G. Wu, Z.-Q. Zhu and T. T. Chu, Chin. Sci. Bull., 1989, 34, 1639 Search PubMed.
- Y. Takaishi, K. Ujita, H. Tokuda, H. Nishino, A. Iwashima and T. Fujita, Cancer Lett., 1993, 68, 129, and references therein Search PubMed.
- Y. Takaishi, S. Ohshima, K. Nakano, T. Tomimatsu, H. Tokuda, H. Nishino and A. Iwashima, J. Nat. Prod., 1993, 56, 815, and references therein Search PubMed.
- Y.-H. Kuo, C.-H. Chen, M.-L. King, T.-S. Wu and K.-H. Lee, Phytochemistry, 1994, 35, 803, and references therein Search PubMed.
- Y. H. Kuo, C. H. Chen, C. F. Chen, M. L. King, H. Y. Chen, K. Chen and K. H. Lee, J. Nat. Prod., 1994, 57, 263 CrossRef CAS.
- Y.-H. Kuo, C.-H. Chen, L.-M. Y. Kuo, M.-L. King, C.-F. Chen and K.-H. Lee, J. Nat. Prod., 1995, 58, 1735, and references therein Search PubMed.
- S. E. Kim, Y. H. Kim and J. J. Lee, J. Nat. Prod., 1998, 61, 108, and references therein Search PubMed.
- J. J. Lee, S. E. Kim and Y. H. Kim, J. Nat. Prod., 1998, 61, 108 CrossRef.
- J. M. Perez-Victoria, B. M. Tincusi, I. A. Jiminez, I. L. Bazzocchi, M. P. Gupta, S. Castanys, F. Gamarro and A. G. Ravelo, J. Med. Chem., 1999, 42, 4388, and references therein Search PubMed.
- X. Y. Li, Int. J. Immunother., 1993, 9, 181, and references therein Search PubMed.
- Y. L. Zheng, Y. Xu and J. F. Lin, Acta Pharm. Sin., 1989, 24, 568 Search PubMed.
- For the only previous review of some of these natural products see: C. H. Heathcock, S. L. Graham, M. C. Pirrung, F. Plavac, C. T. White, in Total synthesis of Natural Products, ed. J. ApSimon, Wiley, New York, 1983, vol. 5, pp. 5–520 Search PubMed.
- H. C. Barrett and G. Büchi, J. Am. Chem. Soc., 1967, 89, 5665 CrossRef CAS.
- X. Chen, S. Shao, T. Li and Y. Li, Synthesis, 1992, 1061 CrossRef CAS.
- J. A. Marshall and M. T. Pike, J. Org. Chem., 1968, 33, 435 CrossRef CAS.
- A. Asselin, M. Mongrain and P. Deslongchamps, Can. J. Chem., 1968, 46, 2817 CAS.
- T. R. Kelly, J. Org. Chem., 1972, 37, 3393 CrossRef CAS.
- C. H. Heathcock and T. R. Kelly, J. Chem. Soc., Chem. Commun., 1968, 267, and references therein Search PubMed.
- G. Zhou, X. Gao, Z. L. Weidong and Y. Li, Tetrahedron Lett., 2001, 42, 3101, and references therein Search PubMed.
- W. Y. Zhang, J. Y. Guo and X. T. Liang, Chin. Chem. Lett., 2000, 11, 873 Search PubMed.
- M. Watanabe and A. Yoshikoshi, J. Chem. Soc., Perkin Trans. 1, 1987, 1793, and references therein Search PubMed.
- D. C. Humber, A. R. Pinder and R. A. Williams, J. Org. Chem., 1967, 32, 2335 CrossRef CAS.
- F. J. Nan, Z. M. Xiong, L. J. Liu, T. S. Li and Y. L. Li, Chin. Chem. Lett., 1995, 6, 1037, and references therein Search PubMed.
- W. J. Xia, L. D. Sun and Y. Q. Tu, Chin. Chem. Lett., 1999, 10, 999, and references therein Search PubMed.
- E. Piers and K. F. Cheng, Can. J. Chem., 1968, 46, 377 CAS.
- Y. Q. Tu and L. D. Sun, Tetrahedron Lett., 1998, 39, 7935, and references therein Search PubMed.
- L. D. Sun, Y. Q. Tu and W. J. Xia, J. Chem. Res., Synop., 1999, 490, and references therein Search PubMed.
- C. Descoins, G. V. Thanh, F.-D. Boyer, P.-H. Ducrot, C. Descoins and J.-Y. Lallemand, Synlett, 1999, 240 CAS.
- F.-D. Boyer and P.-H. Ducrot, Synthesis, 2000, 13, 1868 CrossRef.
- A. C. Spivey, S. J. Woodhead, M. Weston and B. I. Andrews, Angew. Chem., Int. Ed., 2001, 40, 769, and references therein Search PubMed.
- T.-S. Kim and J. D. White, Tetrahedron Lett., 1993, 34, 5535, and references therein Search PubMed.
- W.-D. Z. Li, G. Zhou, X. Gao and Y. Li, Tetrahedron Lett., 2001, 42, 4649 CrossRef CAS.
Footnotes |
| † It has been suggested that maytansine may not be a plant secondary metabolite of the Celastraceae and that it is in fact derived from associated fungi/bacteria.5 |
| ‡ (−)-Dihydroagarofuran is itself a natural product found in galbanum resin and sandalwood oil.4 It is sometimes referred to as (−)-dihydro-β-agarofuran. The β designation here refers to the fact that it is the major isomer obtained by catalytic hydrogenation of (−)-β-agarofuran, which has a double bond between C-4 and C-15.17,18 Similarly, (−)-isodihydroagarofuran (which is epimeric at C-4) is sometimes referred to as (−)-dihydro-α-agarofuran as it is the major isomer obtained by catalytic hydrogenation of (+)-α-agarofuran, which has a double bond between C-3 and C-4.17,18 (+)-α-Agarofuran occurs in agar wood oil.19 |
| § This convention, which engages when the absolute configuration at C-7 is known, places the C-7–C-11 bond in a β-linked orientation, places C-1 as the top left carbon, and labels this ring as the A-ring. The α- and β-face designation and the numbering regime employed (see Fig. 3) are tied to this convention. |
| ¶ It should also be noted that in the numbering regime shown in Fig. 3 the designations of C-14–C-15, and of C-12–C-13 are sometimes interchanged. |
| || An asterix denotes a stereogenic centre for which the absolute configuration from a Celastraceae natural product has yet to be determined. |
| ** The E in cathedulin E-5 designates of Ethiopian origin. Similarly, K and Y designate cathedulins isolated from plants sourced in Kenya and the Yemen Republic, respectively. |
| †† The LD100 is the lethal dose, i.e. that required to kill 100% of the insects. |
| ‡‡ The IC50 is the inhibitory concentration required to kill 50% of cells in vitro. |
| §§ The ED50 is the effective dose required to effect a 50% reduction in viable cells in vivo. |
| ¶¶ The EC50 is the effective concentration required to produce a 50% reduction in viral activity in vivo. |
| |||| The TI is the ratio of IC50 to EC50, and is a measure of the selectivity for diseased cells over healthy cells. |
| *** (−)-10-Epi-α-cyperone has the same absolute stereochemistry at C-7 and C-10 as natural agarofurans. It can be prepared on a large scale in three steps from (−)-carvone by diastereoselective hydrogenation, Robinson annulation (then isomer separation), and dehydration in ∼35% yield.49 |
| ††† Racemisation accompanies conversion of (−)-carvone into (±)-hydroxycarvone because the 1,3-diketo tautomer of (±)-hydroxycarvone is achiral. |
| ‡‡‡ Both the Shapiro (LDA) and Bamford–Stevens (Na, HOCH2CH2OH) conditions have been used previously on related tosylhydrazonyl decalins lacking a hydroxy group at C-4 to give alkenes. The Shapiro conditions give the 2,3-alkene56 whereas the Bamford–Stevens conditions give the 3,4-alkene.57 |
| §§§ (−)-α-Santonin has the absolute configuration depicted in Scheme 9 in which C-10 is epimeric to that required for naturally occurring agarofurans. Consequently, Tu’s products are in the opposite enantiomeric series to that of the natural products. This is clear from the conversion of (−)-α-santonin into (+)-α-cyperone (cf. footnote ***).60 |
| ¶¶¶ It should be noted that in their publications White et al.20 employ the reverse definitions of α-/β-faces and A-/B-rings relative to the those employed here (i.e. as defined in Fig. 3). |
| This journal is © The Royal Society of Chemistry 2002 |