Methods and techniques for the selective extraction and recovery of oxoanions
D. Max
Roundhill
* and
H. Fred
Koch
Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, Texas 79409-1061, USA
First published on 11th December 2001
Abstract
An important goal in environmental chemistry is the extraction of metals that are toxic or radioactive from soils and waters. For many such metals, the problem is solved by designing compounds with coordination sites that are specific for that particular metal. For cases such as oxoanions, however, where the inorganic center is already fully coordinated by oxygens, different strategies need to be used. Chromate, phosphate, selenate, pertechnetate, and aluminate are such anions. These species are problematic contaminants in soils and waters because they are either toxic, environmentally undesirable, or radioactive. Furthermore, under both acidic and basic conditions, the coordination positions of these oxoanions are occupied by oxygens. Chromium(VI) as either chromate or dichromate presents a particular problem because it is a strong oxidizing agent. In this review the different methods of extracting this group of pseudotetrahedral oxoanions are discussed, along with the individual advantages and limitations of each strategy.
 D. Max Roundhill D. Max Roundhill | D. Max Roundhill was born in 1942 in Oldham, England. He studied Chemistry at Oxford University and graduated in 1965. He then moved to Imperial College of the University of London to do his thesis work in the laboratories of Professor Geoffrey Wilkinson. In 1967 he received a PhD degree and moved to Tulane University for postdoctoral research work with Professor Hans B. Jonassen. In 1968 he joined the faculty at Washington State University, where he became Professor of Chemistry. Then in 1983 he moved to Tulane University, and to his present position as Professor in the Department of Chemistry and Biochemistry at Texas Tech University in 1996. His research interests have been in the areas of Organometallics and Homogeneous Catalysis, Inorganic Photochemistry, Coordination Compounds for Imaging, and recently in the design and use of calixarenes for the extraction of metals and oxoanions. |
 H. Fred Koch H. Fred Koch | H. Fred Koch was born in 1966 in Kansas City, Kansas. He received his BS in chemistry from Texas Lutheran College in 1989. He went to Louisiana State University to do his doctoral work for Professor George G. Stanley. In 1996 he received a PhD for work with novel hydroformylation ligand systems. He then moved to Texas Tech University for postdoctoral research work with D. Max Roundhill, studying selective metal extractions. In 1999 he accepted a position with Hoescht Celanese in Corpus Christi, TX. His main research interests have been in the area of metal binding for use in homogeneous catalysis and metal extractions. |
1 Introduction
The extraction and recovery of metals from soils and waters is important if the metal is toxic and of commercial value. In some cases metals can simply be recovered by precipitation, but such methods result in problematic separation steps. A solution phase approach that employs either selective complexant hosts or bioremediation is preferable, especially for ions that are present in aqueous solution. The synthesis of complexants or microbes for specific metals is therefore an important goal for high value toxic metals that need to be selectively recovered from the environment.For inorganic cations there is a wide range of extractants that are available because they can be directly bound to a ligating group on a complexant. However, for inorganic anions such as oxoanions, there is no direct interaction between the inorganic center and the complexant. As a consequence, although numerous molecules act as complexants for cations, few molecules have been identified that act as selective hosts and extractants for anions.1–4 From an environmental viewpoint, a series of anions for which selective hosts would be useful are the oxoanions. Among the important inorganic oxoanions are chromate, phosphate, selenate, pertechnetate, and aluminate. Chromate and dichromate ions are environmentally important due to their high toxicity,5 and also because of their presence in soils and waters from electroplating and other industrial operations.6 At present the remediation of chromate involves reduction to chromium(III), followed by precipitation as its hydroxide. However, engineering considerations cause methods that involve the generation of solid materials to be disfavored. As a result the use of a selective complexant for extracting this reduced state of chromium in soluble form is a preferred alternative when considering approaches for the removal of chromium(VI) from soils and waters. Phosphate is an oxoanion that needs to be removed from soils and waters because it causes excessive plant growth when released into lakes and streams. Also phosphate is widespread in the environment because it is the thermodynamically stable form of phosphorus, and under anerobic conditions all phosphorus compounds are oxidized to phosphates. Under different solution acidities phosphate is present as the different protonated forms H3PO4, H2PO4−, HPO42−, PO43−. Selenium is a toxic element that is found in anionic form in both its selenium(IV) and (VI) oxidation states. In oxygenated alkaline water the higher oxidation state selenate (SeO42−) form predominates, although some selenite (SeO32−) is also present. Since the toxicity of selenium for the two forms is additive, extractants for both forms are needed. Both the pertechnetate (TcO4−) and aluminate oxoanions are special cases since their distribution is localized because they have become an environmental problem as a result of the nuclear industry. Although this article addresses each of the oxoanions, the broader literature on the removal of chromate is reflected in the balance and coverage in the article.
In designing selective extractants for these oxoanions certain strategies are common among them. Each of these anions has peripheral oxygen functionalities that can hydrogen bond with complexant hosts. In addition, each of these anions is present in the form of a salt with a cationic counterion such as sodium or potassium that can be directly bound to ligating groups. It is advantageous therefore to seek extractants that can both coordinate to the cation in the salt and have the correct shape, size, and hydrogen bonding sites to act as a selective host for the oxoanion.
1.1 Toxicological effects
Chromium(VI) is a carcinogen in humans and animals, with chromates and dichromates being both mutagenic and genotoxic. Chromium(VI) requires intracellular reduction for activation, and this in vivo reduction can produce several reactive intermediates such as chromium(V) and chromium(IV) that can target and damage DNA.5 Epidemiological studies have established that chromium(VI) compounds are human carcinogens. They are also strong clastogens, and are mutagenic in both bacterial and mammalian test systems. The principal chemical form of chromium(VI) in solution at physiological pH is chromate, CrO42−, which is isostructural with the other oxoanions considered in this article. Chromate enters mammalian cells through the general anion channel, which results in a rapid accumulation of high concentrations of intracellular chromium. By contrast, water soluble chromium(III) compounds are not considered to be carcinogenic, possibly because they do not cross plasma membranes.7 Nevertheless, the final intracellular reduction product of chromium(VI) is chromium(III) which forms in vivo amino acid nucleotide complexes. The mutagenic potential of these complexes is not fully known. Mussels, which have been widely used as biological monitors of coastal contamination, assimilate chromium.8 Only chromium(VI) from the dissolved phase and chromium(III) from ingested food contribute to chromium accumulation in marine mussels. The uptake of the toxic chromium(VI) is especially important from waters that have low salinities.Selenium is an essential element that is also toxic at higher concentrations. A deficit of selenium leads to white muscle disease in animals, and an excess causes health problems because it replaces sulfur in in vivo biological cycles. From a geological viewpoint, because selenium usually occurs along with sulfur, it is released into the environment from the combustion of high sulfur coal and oil. Pertechnetate, or perrhenate as its surrogate, has become a recent target for the design of extractants because it is a product of nuclear fission. Like the other anions considered in this article, this oxoanion has a tetrahedral structure. However, since it is only singly charged, there is the possibility that it can be selectively extracted in the presence of other more highly charged oxoanions.
1.2 Analytical techniques
The quantity of dissolved oxoanions in water can be determined by several methods. One choice is ion chromatography. This method has detection limits that are in the 0.3–0.4 μg L−1 range. Other methods that have been used include coprecipitation, colorimetry, chelation extraction in combination with either atomic absorption spectroscopy (AAS) or inductively coupled plasma-atomic emission spectroscopy (ICP-AES), and differential pulse polarography.9 Each method has specific features that makes it preferable in certain situations and conditions. Phosphate and aluminate can also be selectively determined by 31P and 27Al NMR spectroscopy respectively, although the sensitivity of this method is low. The ICP-AES method also has a rather low sensitivity for phosphate. Technetium can be easily assayed from its radioactive signature.2 Extraction methods
Several approaches have been used for the environmental removal of oxoanions. These approaches involve precipitation methods as well as electrochemical and complexation methods. Both liquid–liquid phase extraction and ion exchange are widely used methods. In designing selective extractants, the function of the material both as a complexant and a phase transfer agent must be considered for maximum effectiveness.2.1 Precipitation methods
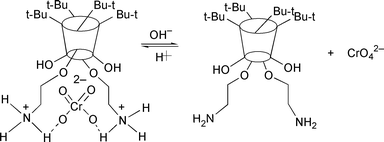
Precipitation methods have been widely used for the removal of chromium because its toxic hexavalent form is a strong oxidizing agent that is destructive to many complexants. Conventional methods for chromium recovery involve reduction of chromium(VI) to chromium(III), followed by precipitation of the chromium(III) hydroxide Cr(OH)3 at a solution pH in the 8–10 range. One method that is used to technologically achieve this goal is to reduce the chromium(VI) in the presence of oxide surfaces with α-hydroxycarboxylic acids such as mandelic acid. Alternatively, either oxalic acid or substituted phenols can be used as the reductant. These conventional pump-and-treat techniques for chromium(VI) removal, however, frequently result in lengthy application times because of the decrease in the chromium removal rate at the later stages of the process. Nevertheless, modifications can be made to improve the process. One improvement involves the addition of sulfate ion to the extraction water, which decreases by almost two orders of magnitude the number of pore volumes required to achieve a targeted level of chromium(VI) reduction. This effect is a consequence of sulfate releasing chromate from any insoluble barium chromate according to eqn. (1).| BaCrO4 + SO42− → BaSO4 + CrO42− | (1) |
Phosphate can also be recovered in solid form if desired because under high pH conditions its trianionic form gives very insoluble salts with cations such as aluminum(III) and calcium(II).
2.2 Electrochemical methods
Electroplating baths are a source of waste chromium, and an electrochemical precipitation (ECP) process has been used to remove chromium from electroplating wastewater. The chromium removal efficiencies by such methods are greater than 99%, and the residual chromium concentrations are less than 0.5 mg L−1. The ECP process uses a current of 0.5–5.0 amperes and an initial pH of 4.5.2.3 Soil washing
Hexavalent chromium can be extracted directly from soils by using a mixture of sodium carbonate and sodium hydroxide in the temperature range of 90–95 °C. Extraction with 0.28 M sodium carbonate and 0.5 M sodium hydroxide solutions results in maximal dissolution of chromium(VI), while minimizing oxidation and reduction.13 Ambient temperature mixed solutions containing sodium carbonate and sodium hydroxide, pure water itself, a phosphate buffer at pH 7.0, or sonication under alkaline conditions, all extract lesser amounts of chromium(VI). Other methods such as ion exchange or adsorption, both of which use solid phase materials, have also been successfully used to remove chromium(VI). Examples of adsorbents that have been used are anthracite and activated carbon. A mixture of iron(III) and chromium(III) hydroxide has also been used as an adsorbent for chromium(VI), with almost complete removal of chromium(VI) being achieved from water that is in the pH range of 5–6. Nevertheless, effective reliable methods for extracting both soluble and insoluble forms of chromium(VI) from soils without inducing chromium(III) oxidation or chromium(VI) reduction are still required.2.4 Extraction with minerals
In addition to these prepared materials, minerals can also be used as adsorbents for chromium(VI). One such mineral is goethite (α-FeOOH).14,15 An analysis of the structural EXAFS data concludes that the three different surface complexes of chromate that are formed on goethite are a monodentate complex 1, and both a mononuclear 2 and a binuclear 3 bidentate complex. Pressure jump relaxation measurements on the system show that the adsorption of chromate on goethite is a two-step process that results in the formation of an inner-sphere bidentate surface complex. The initial rapid step involves a ligand exchange reaction of the protonated aqueous form (HCrO4−) with hydroxy groups on the goethite surface to give an inner-sphere monodentate surface complex. Zeolites have also been used as adsorbents for chromium(VI).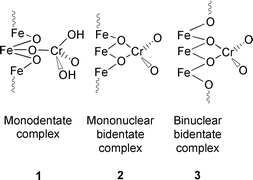
2.5 Polymeric sorbents
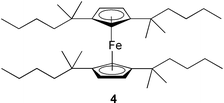
A polymeric ligand exchange material has been developed that removes trace concentrations of chromium(VI), even in the presence of sulfate, chloride, or bicarbonate ions. The material consists of a cross-linked polystyrene–divinylbenzene matrix with picolylamine groups appended. Onto these picolyl groups is chelated copper(II), and the high selectivity results from the preference of chromium(VI) for the divalent copper center.16 Chromium(III) has been removed from phosphoric acid solutions by chelating resins containing either phosphonic or diphosphonic groups. One such resin is Diphonix that contains both sulfuric and gem-diphosphonic acid groups, and effectively removes chromium(III). The column sorption is influenced by the acid concentration, with a decrease in elution efficiency of chromium(III) from the Diphonix being obtained with increases in phosphoric acid concentration during the sorption process.17 The iron(III) complex of a carboxylated polyacrylamide-grafted sawdust is also an effective adsorbent for the removal of chromium(VI) from aqueous solution. Up to 99% removal is observed in the pH range of 2.0 to 3.0. The chromium(VI) adsorbs as HCrO4−. Over 95% of the chromium(VI) can be desorbed with 0.1 M sodium hydroxide solution.18 An alkyl quaternized poly(4-vinylpyridine) coated silica gel absorbs chromium(VI) from solution at a diffusion controlled rate.19 Chromium(VI) has also been removed using polyurea microcapsules.For a series of different ion exchange resins it is found that the acidities must be carefully chosen. Because of selectivity reversal between HCrO4− and CrO42− at the prevailing ionic strength there is a critical pH in the acidic region whereby lowering it causes no increase in chromate removal capacity. An ion exchange material prepared by adsorbing a ferrocenyl salt onto silica is a redox-recyclable extractant for pertechnetate and perrhenate. The oxidized cationic form of this compound, 1,1′,3,3′-tetrakis(2-methyl-2-hexyl)ferrocene 4, extracts these anions into an organic phase and then releases them upon reduction with potassium ferrocyanide. The reversibility of this material results from the oxidized iron(III) form being cationic, and the reduced iron(II) form being uncharged.20
3 Complexant design
Although to date there has been relatively little effort directed toward designing selective receptors for oxoanions, the situation is rapidly changing. Simple compounds that have been used as liquid–liquid extractants for chromium(VI) are methyl isobutyl ketone, p-(1,1,3,3-tetramethylbutyl)phenylphosphoric acid, an iron(III) hexamethylenedithiocarbamate complex, and diphenylcarbazide. Selectivity, however, is often limited, and there is no rational understanding as to why they are effective. As a result, no guidelines are available as to how to design complexants and extractants that have high selectivities for chromate or other oxoanions. An important aspect of developing extractant for both metal ions and oxoanions is that they are kinetically rapid. This is especially important if the complexant will eventually be bound to a polymeric support because attachment of a chelate to such a support results in a decrease in the rate of metal binding.3.1 Cationic amines
One approach to specifically designing extractants for oxoanions is to seek cationic hosts with tetrahedral cavities that can hydrogen bond with the peripheral oxygens on the guest. These cations can have metal centers that bind to the oxo functionalities of the anions or cationic groups that can interact by hydrogen bonding. Amines meet many of these requirements, and reversibility of anionic binding can be achieved by switching between the free amine and its protonated form.In acid solution the chromium(VI) oxoanion is primarily present in its protonated dimeric form HCr2O7−. Extraction from acid solution via ion-pair formation with simple long-chain alkylamines R3N results in proton transfer to the amine [eqn. (2)].
| HCr2O7− + H+ + 2R3N → (R3NH)2Cr2O7 | (2) |
| 2Na+ + CrO42− + 2R4N+Cl− → 2Na+ + 2Cl− + 2(R4N)2CrO4 | (3) |
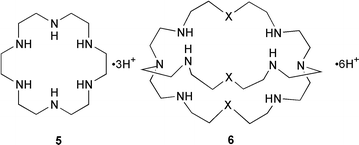
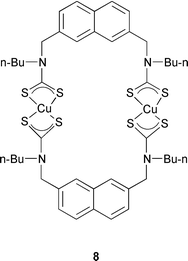
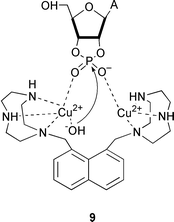
A water-soluble polyazaferrocene macrocycle 7 has been employed as an oxoanion sensor by again making use of the reversible 1-electron oxidation wave of a ferrocenyl reporter. This compound electrochemically recognizes both the hydrogen phosphate HPO42− and ATP oxoanions in an aqueous environment in the pH range of 6 to 7.25 Metallocycles are also used for the recognition of oxoanions. The metallocycle 8 selectively recognizes both the perrhenate and dihydrogen phosphate oxoanions. The anion selectivity of 8 is influenced by the size of the dithiocarbamate copper(II) macrocycle.26 A metallocrown dimer that contains five tyrosine-hydroximate ligands and five copper(II) ions is a selective host for nitrate anions. This host–guest complexation is a good size match because it occurs with only minimal perturbations to the cavity geometry.27 A similar host–guest strategy uses a dicopper(II) cationic complex within a polyamine cleft 9 for binding to phosphoryl groups.28
3.2 Calixarenes as extractants
Calixarenes, which are cyclic oligomers prepared by condensation reactions between para substituted phenols and formaldehyde, are excellent phase transfer agents. Calix[4]arenes have both a wide and narrow rim. They have tert-butyl groups on the wide rim and phenol functionalities on the narrow one. Two abbreviations that are used for calix[4]arenes are shown in Fig. 1.![Structural representations for a calix[4]arene.](/image/article/2002/CS/b003141k/b003141k-f1.gif) | ||
| Fig. 1 Structural representations for a calix[4]arene. | ||
| (4) |
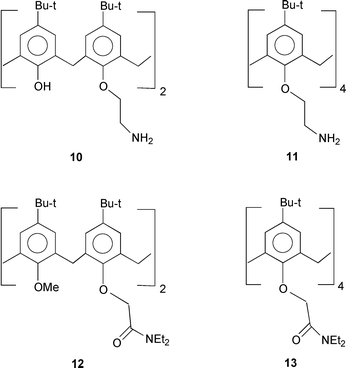
Chemically modified calix[4]arenes are effective extractants for transferring cationic metal ions from aqueous solution into an organic layer.29,30 Since polyamines are hosts for oxoanions it is logical to target calixarenes with multiple amine functionalities as oxoanion extractants. As a result a calix[4]arene diamine has been found to be a pH-switchable extractant for chromium(VI). The diprotonated alkylammonium form transfers dichromate into an organic phase, but at a higher pH the chromate ion is carried back into the aqueous phase [eqn. (4)].31 A limitation to amines is that they are susceptible to oxidation by chromium(VI). Amides, however, are more resistant to oxidation, therefore the analogous amides have also been used as extractants. These calix[4]arene amines 10, 11 and amides 12, 13 show significant liquid–liquid extraction of oxoanions from water into a chloroform phase.21 The extraction percentages in Table 1 show that 11 extracts all oxoanions, and that 13 is effective for perrhenate. The amides, however, have the advantage in that they have both amide nitrogen and carbonyl oxygen functionalities for hydrogen bonding with the oxoanion, a feature that may be a factor in the extraction of selenate by hydrophobic calix[4]arene amides.32 Since these extractions are carried out for only a short time duration the complexation and phase transfer sequence is relatively rapid. Longer contact times cause no significant changes in the extraction percentages, which indicates that equilibration is quickly reached. Pyridinium substituted calixarenes are also hosts for both dihydrogen phosphate and hydrogen sulfate oxoanions.33
| Compound | Cr2O72− | CrO42− | HPO42− | ReO4− | WO42− |
|---|---|---|---|---|---|
| 10 | 11 | 9 | — | — | 10 |
| 11 | 16 | 88 | 66 | 36 | 95 |
| 12 | — | 6 | 3 | 1 | 1 |
| 13 | 1 | 5 | — | 29 | 4.5 |
Other cationic calixarenes have been used as extractants by ion-pairing with oxoanions. One such family of compounds incorporates either a cobalt(III) or iron(II) metallocene (14–17), or a ruthenium(II) bipyridyl cation 18 bound to the wide calixarene rim. In addition to having a cationic metal center, these extractants again have amide functionalities that can become involved in hydrogen bonding with the oxoanion guest. The stability constant data for 15–17 with oxoanions and the chloride are collected in Table 2. A high selectivity for the dihydrogen phosphate oxoanion is observed. As with the data in Table 2, it is clear that oxoanions associate with these calixarenes, but it is difficult to rationalize the observed selectivities. In addition to functioning as extractants for oxoanions, these compounds also act as either redox or optical sensors for oxoanions.1–3
| Compound | H2PO4− | HSO4− | NO3− | Cl− |
|---|---|---|---|---|
| 15 | 2500 | — | — | 400 |
| 16 | 3100 | weak | weak | weak |
| 17 | 6380 | 40 | 125 | 70 |
3.3 Poly(ethylene glycol) for chromium(VI) extraction
An aqueous solution of poly(ethylene glycol) (PEG) is a biphasic system that has been used as a medium for the phase transfer recovery of metals from salt solutions. When ammonia is bubbled through an aqueous solution having the chromium(VI) in the upper PEG-rich layer, the chromium is transferred to the lower layer. Similarly, passing carbon dioxide through the system in aqueous ammonia with chromium(VI) in the lower layer results in its transfer to the upper layer. Subsequent addition of ferrous sulfate leads to reduction of chromium(VI) to chromium(III), followed by an essentially quantitative transfer of the chromium from the PEG to the salt layer. A combination of electrolytic methods coupled with the iron(II,III) redox system can be used to reduce chromium(VI) to chromium(III). The system succeeds because after oxidation of the added iron(II) to iron(III) by chromium(VI), the iron(III) is electrolytically reduced back to iron(II). The iron(II), therefore, needs to be present in only catalytic quantities.343.4 Supercritical carbon dioxide for rhenium(VI) extraction
Supercritical carbon dioxide is a hydrophobic fluid that is attracting increasing attention for metal extractions. Oxoanions are also being studied. Perrhenate as its tetrabutylammonium salt forms the most efficient ion pair for maximizing extraction into a solution of methanol in supercritical carbon dioxide. The salt is extracted because the charge on the perrhenate anion is partially neutralized by electrostatic interactions of the ion pair, thereby making the salt soluble in the hydrophobic carbon dioxide solvent. Increasing or decreasing the length of the alkyl chain decreases the extraction efficiency.354 Extraction from soils
Removal of metals from soil presents different challenges than does their removal from water. In particular, it is economically advantageous to effect the in situ remediation of soils without having to carry out their excavation and removal for off-site cleansing. This is also a particularly important detail if the soil is covered by buildings or vegetation that needs to be retained. The analytical challenges can also be problematic. One reason is that the metal may be strongly occluded into the zeolite soil structure, and therefore difficult to release for accurate assay. Nevertheless, chromium(VI) has been successfully removed from soils. The oxidation–reduction chemistry of chromium is an important factor in determining remediation strategies for soils. Certain forms of chromium(III) can be oxidized to chromium(VI), and also chromium(VI) can be reduced to chromium(III), under differing soil conditions. It has been proposed that a Potential Chromium Oxidation Score (PCDOS) be used as the basis for setting acceptable maximum limits for chromium(III)-containing wastes in particular soil environments to maintain chromium(VI) levels at or below health-based limits. This PCDOS is based on solubility and site potential of chromium(III), levels of manganese(III) and manganese(IV), soil potential for chromium(VI) reduction, and soil pH.36 Much of the prior engineering work on in situ soil reclamation has focused on the removal of organic compounds by using technologies developed for the recovery of oil through polymer and/or surfactant flooding of porous media. Many of these strategies, however, can also be used for metals such as chromium.374.1 Soil washing
The use of flooding methods for the removal of toxic metals from soils by washing with highly alkaline or acid water has been investigated, but to a lesser extent than for organics. Chromium has been effectively removed from soil by using a washing solution at a pH of 10.4. A similar in situ application of soil washing uses an acid solution to leach metals from more than 30000 cubic meters of soil.384.2 In situ stabilization
Another approach for dealing with metal contamination in soils is in situ stabilization. The in situ stabilization involves converting the metal to an insoluble form such as an insoluble sulfide, thereby converting the metal into the same chemical composition as the mineral from which it was originally extracted. Another alternative involves solidification and stabilization into Portland cement prior to burial and disposal. An agricultural-based alternative is also viable. Such a method uses green plants and soil amendments to achieve in situ stabilization.395 Electrokinetic extraction
The direct removal of metals such as anionic chromium(VI) from soils without introducing chemicals presents another challenge. A technique that can be used for metals is electrokinetic extraction. The electrokinetic method involves inserting electrodes into the soil and then applying a potential across them. In this technique the application of an in situ direct current produces an electroosmotic water flow, and the H+ and OH− fronts move through the soil in opposite directions.40 The acid front that moves from the anode to the cathode dissolves metals that are absorbed in the soil. By contrast, the highly alkaline interstitial water near the cathode causes these heavy metals to precipitate as their hydroxides. This problem limits the extent of metal ion migration, and hence, to some extent, the effectiveness of the electrokinetic reclamation process (Fig. 2). Electrokinetic remediation has been used to treat chromium(VI) contaminated soils. Complete cleansing of a sample of 50–100 mesh sand containing 10% water and 100 ppm chromium(VI) is obtained after 22 hours of treatment. The chromium(VI) that collects near the anode migrates at a rate of 0.40 cm h−1 with a pure water current density of 2.26 mA cm−2. The results of such a homogeneous run with chromate contaminated sand are shown in Fig. 3. During the run the current remains constant at 10.3 mA, while the the voltage rises from the initial value of 90 V to a final value of 150 V after 22 hours. No chromium is detected in the sample except in a region adjacent to the anode. These studies indicate that the electromigration rates normalized by pure water current densities in homogeneous studies can also be applied to heterogeneous soil systems. The in situ electrokinetic remediation of chromium(VI) is found to be dependent on the type of clay soil onto which it is adsorbed. In a comparative study of kaolin clay and a glacial till, some differences were found. In the kaolin a sharp pH gradient ranging from ∼2 near the anode to 12 near the cathode develops. However, in the glacial till, alkaline conditions (pH > 8) exist throughout the soil because of its high carbonate-buffering capacity. The migration of anionic chromium(VI) toward the anode is therefore more efficient in the glacial till, and also near the cathode region of kaolin, because of low adsorption of chromium(VI) in high pH environments. Adsorption of chromium(VI) in the low pH regions near the anode in kaolin results in low chromium(VI) removal.41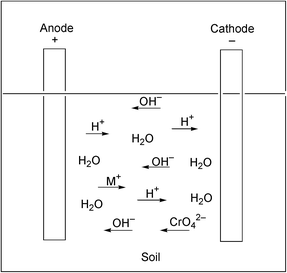 | ||
| Fig. 2 Electrokinetic transfer of the chromate oxoanion and electroosmosis in soils | ||
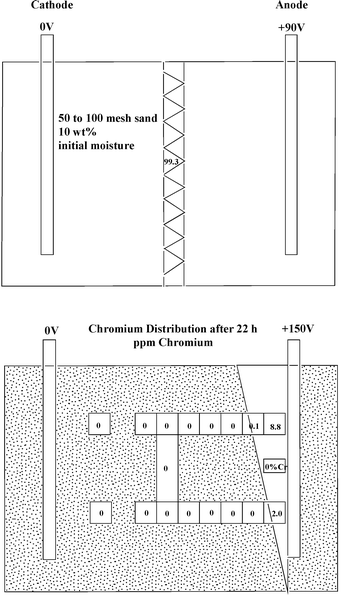 | ||
| Fig. 3 Chromium(VI) distribution after electrokinetic treatment. | ||
6 Bioremediation
Bioremediation is a technology that differs from the simple use of biomass for the adsorption of metals such as chromium.42,43 Instead, it is a technology that focuses on developing problem-specific organisms. As a result, bioremediation, which uses living microorganisms for the degradation of pollutants, is becoming increasingly important in the search for a clean environment. Furthermore, genetically engineered microbes can be designed that target specific pollutants and environmental conditions. Although bioremediation is primarily used for organic pollutants, there are also possibilities for the development of biotechnologies that are focused on the remediation of oxoanions such as chromate. As an example, Allied Signal has developed a process for the bioremediation of groundwater and soils contaminated by metals. The process is successful for chromium(VI). The process takes advantage of the ability of anaerobic sulfate-reducing bacteria to reduce metals to their lower oxidation states, which for chromium(VI) leads to its conversion to the less toxic chromium(III) form. The process circumvents the acute toxicity of chromium(VI) to microbes by using bacteria that produce hydrogen sulfide gradients, with the chromium(VI) being reduced to chromium(III) before it reaches the microbial cells. This technology has already been successfully field tested.An alternate approach for chromium remediation couples the microbial reduction of chromium(VI) with the anaerobic degradation of benzoate.44 Chromium(VI) is removed from aqueous solutions by an Enterobacter cloacae strain that reduces it under anaerobic conditions. Both chromium and organic contaminants can be simultaneously removed by an anaerobic consortium of bacteria. The microbial reduction of chromium(VI), and its potential application for the removal of chromium from polluted sites, has been reviewed.45 Polluted sites often contain mixtures of both chromium(VI) and toxic organic compounds because industrial processes such as leather tanning, metals finishing, and petroleum refining, discharge mixtures of chromium(VI) and aromatics. An important goal, therefore, is the simultaneous detoxification of both chromium(VI) and these phenolic compounds using aerobic bacteria or abiotic redox reactions.46 Chromium(VI)-resistant fungal or bacterial microbes can also be used to effect the reduction of chromium(VI) to chromium(III). In comparative experiments, it has been found that the fungi tested are more tolerant to chromium(VI) than are the bacteria, and that all microbes are more effective reductants under anaerobic conditions. In order to be able to apply these technologies a two-stage bioreactor has been designed where Escherichia coli cells are grown in a first stage, and then pumped to a second stage plug-flow reactor where the anaerobic reduction of chromium(VI) occurs.47
7 Phytoremediation
Phytoremediation is also an important method for the removal of metals and oxoanions from soils and waters. For example, the efficiency of wheat, rape and buckwheat for the removal of chromium from waste waters has been compared, with buckwheat being the most efficient. The plant roots are powerful agents for chromium(III) removal, inducing its subsequent precipitation on their surface. Although the chromium(VI) form is more toxic to plants than is the chromium(III) form, some plants retain their capacity for chromium removal while still showing symptoms of strong toxicity. Chromium(VI) is removed from aqueous solutions by both Zoogloea ramigera and water hyacinth.8 Conclusions and perspectives
This field of anion recognition and extraction is a relatively new one, but one that has far reaching consequences. The field of oxoanions is a particularly important one because of environmentally problematic ions such as chromate, phosphate, pertechnetate and selenate. Methods of extraction of the anions from soils and waters are becoming more readily available, however, and systems are being discovered that can select between these oxoanions and the simple halides. Nevertheless, no chemical system is sufficiently effective at this time to be the chosen technology. However, different systems will continue to be introduced until all of the precipitation methods have been replaced.In competition with the chemical methods are those of bioremediation and phytoremediation. These last two methods have the advantage of not introducing chemicals into the soil or water that must be subsequently removed. Also the two methods are complementary to some extent in that it is becoming apparent that the metal binding sites in bioremediation and phytoremediation systems are chemically similar to those that are designed into selective ligating sites. Advances in genetic engineering will allow for systems to be designed and grown that have increasingly higher capacities and selectivities. Successful remediation strategies will likely be ones that take advantage of all of the methods in order to engineer the most effective system. Nevertheless, changing site conditions will always require local variations in the chosen method.
9 Acknowledgments
We thank the U.S. Army Research Office, the Robert A. Welch Foundation, and the U.S. Department of Energy, through the Pacific Northwest National Laboratory, for support of our research on metal extraction with calixarenes.10 References
- D. M. Roundhill, Metal Extraction from Soils and Waters, Kluwer Academic/Plenum Publishers, New York, NY, 2001 Search PubMed.
- Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic, Dordrecht, 2001 Search PubMed.
- P. D. Beer, Acc. Chem. Res., 1998, 31, 71 CrossRef CAS.
- F. C. J. M. Van Vagile, W. Verboom and D. N. Reinhoudt, Chem. Rev., 1994, 94, 281.
- D. M. Stearns, L. J. Kennedy, K. D. Courtney, P. H. Giangrande, L. S. Phieffer and K. E. Wetterhahn, Biochemistry, 1995, 34, 910 CrossRef CAS.
- P. R. Wittbrodt and C. D. Palmer, Environ. Sci. Technol., 1995, 29, 255 CAS.
- A. Zhitkovich, V. Voitkun and M. Costa, Biochemistry, 1996, 35, 7275 CrossRef CAS.
- W.-X. Wang, S. B. Griscom and N. S. Fisher, Environ. Sci. Technol., 1997, 31, 603 CrossRef CAS.
- W. Mueller and D. L. Smith, Compilation of E.P.A.’s Sampling and Analysis Methods, ed. L. H. Keith, 1991, Lewis Publishers Inc., CRC Press Inc., Boca Raton, FL Search PubMed.
- L. D. Skrylev, T. L. Skryleva and A. N. Purich, J. Water Chem. Technol., 1996, 18, 25 Search PubMed.
- Y. El-Shoubary, N. Speizer, S. Seth and H. Savoia, Environ. Progr., 1998, 17, 209 Search PubMed.
- B. Wielinga, M. M. Mizuba, C. M. Hansel and S. Fendorf, Environ. Sci. Technol., 2001, 35, 522 CrossRef CAS.
- B. R. James, J. C. Petura, R. J. Vitale and G. R. Mussoline, Environ. Sci. Technol., 1995, 29, 2377 CrossRef CAS.
- S. Fendorf, M. J. Eick, P. Grossl and D. L. Sparks, Environ. Sci. Technol., 1997, 31, 315 CrossRef CAS.
- P. Grossl, M. J. Eick, D. L. Sparks, S. Goldberg and C. C. Ainsworth, Environ. Sci. Technol., 1997, 31, 321 CrossRef CAS.
- D. Zhao, A. K. SenGupta and L. Stewart, Ind. Eng. Chem. Res., 1998, 37, 4383 CrossRef CAS.
- N. Kabay, M. Demircioǧlu, H. Ekinci, M. Yüksel, M. Saǧlam, M. Akçay and M. Streat, Ind. Eng. Chem. Res., 1998, 37, 2541 CrossRef CAS.
- M. R. Unnithan and T. S. Anirudhan, Ind. Eng. Chem. Res., 2001, 40, 2693 CrossRef CAS.
- D. Gang, W. Hu, S. K. Banerji and T. E. Clevenger, Ind. Eng. Chem. Res., 2001, 40, 1200 CrossRef CAS.
- C. K. Chambliss, M. A. Odom, C. M. L. Morales, C. R. Martin and S. H. Strauss, Anal. Chem., 1998, 70, 757 CrossRef CAS.
- N. J. Wolf, E. M. Georgiev, A. T. Yordanov, B. R. Whittlesey, H. F. Koch and D. M. Roundhill, Polyhedron, 1999, 18, 885 CrossRef CAS.
- Z.-F. Yang, A. K. Guha and K. K. Sirkar, Ind. Eng. Chem. Res., 1996, 35, 1383 CrossRef CAS.
- J. S. Bradshaw, K. E. Krakowiak and R. M. Izatt, Tetrahedron, 1992, 48, 4475 CrossRef CAS.
- B. Dietrich, Pure Appl. Chem., 1993, 65, 1457 CrossRef CAS.
- P. D. Beer, Z. Chen, M. G. B. Drew, J. Kingston, M. I. Ogden and P. Spencer, J. Chem. Soc., Chem. Commun., 1993, 1045 Search PubMed.
- P. D. Beer, N. Berry, M. G. B. Drew, D. Fox, M. E. Padilla-Tosta and S. Patell, Chem. Commun., 2001, 199 RSC.
- A. D. Cutland, R. G. Malkani, J. W. Kampf and V. L. Pecoraro, Chem. Commun., 2001, 199 RSC.
- P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Chem. Soc. Rev., 2000, 29, 75 RSC.
- D. M. Roundhill, Prog. Inorg. Chem., 1995, 43, 533 Search PubMed.
- A. T. Yordanov, J. T. Mague and D. M. Roundhill, Inorg. Chem., 1995, 34, 5084 CrossRef CAS.
- E. M. Georgiev, N. Wolf and D. M. Roundhill, Polyhedron, 1997, 16, 1581 CrossRef CAS.
- O. M. Falana, H. F. Koch, G. Lumetta, B. Hay and D. M. Roundhill, Chem. Commun., 1998, 503 RSC.
- P. D. Beer, M. G. B. Drew and K. Gradwell, J. Chem. Soc., Perkin Trans 2, 2000, 511 RSC.
- H. F. Koch, J. Shen and D. M. Roundhill, Sep. Sci. Technol., 2000, 35, 623 Search PubMed.
- J. A. Gawenis, J. F. Kauffman and S. S. Jurisson, Anal. Chem., 2001, 73, 2022 CrossRef CAS.
- B. R. James, J. C. Petura, R. J. Vitale and G. R. Mussoline, J. Soil Contam., 1997, 6, 569 Search PubMed.
- A. S. Abdul and T. L. Gibson, Environ. Sci. Technol., 1991, 25, 603.
- T. H. Pfeifer, T. J. Nunno and J. S. Walters, Environ. Prog., 1990, 9, 79 Search PubMed.
- W. R. Berti and S. D. Cunningham, Environ. Sci. Technol., 1997, 31, 1359 CrossRef CAS.
- R. Lageman, W. Pool and G. Seffinga, Chem. Ind., 1989, 585 Search PubMed.
- K. R. Reddy and V. S. Parupudi, J. Soil Contam., 1997, 6, 391 Search PubMed.
- J. M. Chen and O. J. Hao, J. Chem. Tech. Biotech., 1997, 69, 70 Search PubMed.
- D. Kratochvil, P. Pimentel and B. Volesky, Environ. Sci. Tech., 1998, 32, 2693 Search PubMed.
- H. Shen, P. H. Pritchard and G. W. Sewell, Environ. Sci. Technol., 1996, 30, 1667 CrossRef CAS.
- D. R. Lovley, Ann. Rev. Microbiol., 1993, 14, 158 Search PubMed.
- M. S. Elovitz and W. Fish, Environ. Sci. Technol., 1994, 28, 2161 CAS.
- H. Shen and Y-T. Wang, J. Environ. Eng., 1995, 121, 798 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |