Infrared chemiluminescence: Evidence for adduct formation in the H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) +CH2XI reaction and studies of the N+CH2X (X=Cl/F/I/H) reactions
+CH2XI reaction and studies of the N+CH2X (X=Cl/F/I/H) reactions
Received
14th September 2001
, Accepted 26th October 2001
First published on 6th December 2001
Abstract
Infrared chemiluminescence from a flow reactor has been used to study the H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +CH2XI and N+CH2X (X=Cl, F, I, H) reactions at 300 K. Both the HI+CH2Cl and HCl+CH2I channels were identified for the H+CH2ClI reaction. The HCl channel involves adduct, HICH2Cl, formation as confirmed by the D+CH2ClI reaction, which gave both HCl and DCl products. The nascent HCl(v) distribution from the H+CH2ClI reaction was P1–P5=25 : 29 : 26 : 13 : 7. The rate constant for the HCl(v) formation channel is estimated to be 4 times smaller than that for the H+Cl2 reaction. The highest HCl(v) level observed
from the H+CH2ClI reaction implies that the C–Cl bond energy is 50.2 kJ mol−1 lower than that of the Cl–CH3 bond, which is in modest agreement with recent theoretical estimates. The H+CH2FI reaction gave a HF(v) distribution of P1–P3=77 : 15 : 8. The C–F bond energy in CH2FI is estimated to be ≤460.2 kJ mol−1, based on the highest HF(v) level observed, the upper bound being the same as that of F–CH3. When N atoms are added to the flow reactor, the HCl(v) emission intensities from H+CH2ClI increased by up to 2-fold, which is attributed to the N+CH2Cl→HCl+HCN reaction. Concomitant weak emission from HCN and HNC could
also be observed; however, the main product channel is thought to be NCH2+Cl. Strong visible CN(A–X) emission was also observed when H/N/CH2XI were present in the reactor. If the CH2X radicals were produced by the F+CH3X reaction in the presence of N atoms, similar results were obtained. The N+CH2N reaction is proposed as the first step that leads to CN(A) formation with NCN as an intermediate.
+CH2XI and N+CH2X (X=Cl, F, I, H) reactions at 300 K. Both the HI+CH2Cl and HCl+CH2I channels were identified for the H+CH2ClI reaction. The HCl channel involves adduct, HICH2Cl, formation as confirmed by the D+CH2ClI reaction, which gave both HCl and DCl products. The nascent HCl(v) distribution from the H+CH2ClI reaction was P1–P5=25 : 29 : 26 : 13 : 7. The rate constant for the HCl(v) formation channel is estimated to be 4 times smaller than that for the H+Cl2 reaction. The highest HCl(v) level observed
from the H+CH2ClI reaction implies that the C–Cl bond energy is 50.2 kJ mol−1 lower than that of the Cl–CH3 bond, which is in modest agreement with recent theoretical estimates. The H+CH2FI reaction gave a HF(v) distribution of P1–P3=77 : 15 : 8. The C–F bond energy in CH2FI is estimated to be ≤460.2 kJ mol−1, based on the highest HF(v) level observed, the upper bound being the same as that of F–CH3. When N atoms are added to the flow reactor, the HCl(v) emission intensities from H+CH2ClI increased by up to 2-fold, which is attributed to the N+CH2Cl→HCl+HCN reaction. Concomitant weak emission from HCN and HNC could
also be observed; however, the main product channel is thought to be NCH2+Cl. Strong visible CN(A–X) emission was also observed when H/N/CH2XI were present in the reactor. If the CH2X radicals were produced by the F+CH3X reaction in the presence of N atoms, similar results were obtained. The N+CH2N reaction is proposed as the first step that leads to CN(A) formation with NCN as an intermediate.
I. Introduction
This paper reports the results of infrared chemiluminescence (IRCL) studies of the H+CH2XI and N+CH2X (X=Cl/F/I/H) reactions. Our initial interest in the H+CH2ClI reaction was to use it to produce CH2Cl in the flow reactor for subsequent studies of the N+CH2Cl reaction. Iodine atom abstraction reactions by H atoms are usually fast and the Kansas State Laboratory has successfully used the reaction of iodoalkanes with H atom as precursor for various radicals.1–4 For example, the rate constants for the H+CF3I/CH3I reactions5–7 are both about 1×10−11 cm3 molecule−1 s−1. As
the investigation proceeded, it was realized that the primary reaction, H+CH2ClI, was interesting and needed to be characterized on its own merit. The chemistry of iodine-containing molecules is important in the earth's atmosphere.8 Major sources for I atoms in the atmosphere are CH3I and CH2ClI.8 Photodissociation of these iodocompounds9,10 and their reactions with Cl11,12 and O13 atoms have been studied as possible sinks for CH2ClI in the atmosphere. As pointed out above, H atoms rapidly react with iodoalkanes and the H atom reaction could also be an important sink for CH2ClI. No study of this reaction has yet been reported, to the best of our knowledge.
Reactions of O atoms13 and Cl atoms12 with CH2ClI appear to proceed via bound intermediates. Leone and co-workers14 have shown that the mechanism for O+C2H5I and larger alkyl iodides involves a bound intermediate that gives HOI. Reversible adduct formation has also been observed for Cl+CH3I15 and F+CH2BrCl.16 For reactions of H atoms with IX compounds (X=Cl/F), an H migration step following adduct formation has long been recognized.17–19 In the present study infrared chemiluminescence from HCl(v) was observed from the H+CH2ClI reaction, and an adduct seems to facilitate
HCl formation. At least two channels are possible:
| |  | (R1) |
| |  | (R2) |
| | 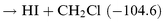 | (R1) |
The thermochemistry is not well established, but the enthalpies of reaction (in kJ mol
−1) based upon our thermochemical estimates,
vide infra, are given in parentheses. The HI formation could be either direct or
via the adduct, but the HCl formation is thought to proceed only through adduct formation. Direct Cl abstraction from
chloroalkanes has a large activation barrier and this reaction has not been observed at 300 K in a flow reactor.
2 The D
+
CH
2ClI and H/D
+
CH
2FI reactions were also investigated to elucidate the adduct formation mechanism.
Our interest in the N+CH2Cl reaction arose because of the importance of the N+CH3 reaction in planetary atmospheres and combustion processes.20–25 For this reaction, the product channels have been actively discussed.
| |  | (R3) |
| |  | (R4) |
| |  | (R5) |
The N
+
CH
3 reaction was proposed to account for the presence of HCN in Titan's atmopsphere.
20 However, laboratory studies of this reaction showed that the most exothermic channel,
(R4), is not the dominant channel.
22 In fact, the CH
2N
+
H channel accounts for 90%
of the reaction and the HCN
+
H
2
(or 2H) channel is estimated to contribute ≈
10%. The HCN can also be formed by the disproportionation reactions, H
+
CH
2N
→
HCN
+
H
2 or N
+
CH
2N
→
HCN
+
NH.
23 The N
+
CH
3 reaction presumably proceeds by recombination giving the triplet methyl nitrene radical, which then dissociates. The triplet CH
3N radical (X
3A
2) requires a spin change to give HCN
+
H
2. Sadygov and Yarkony
20 evaluated the intersystem crossing between the CH
3N (X
3A
2) and (1
1A
′) states and identified a zero-barrier path for
(R4). Due to the large exoergicity, the HCN product could be vibrationally excited. As far as we know, no attempt has been made to look for HCN by IRCL from this reaction. HCN infrared chemiluminescence, which is relatively easy to observe, has been used to study the dynamics of several reactions.
26–28
It is well known that unimolecular H2 elimination processes from ground state molecules have high barriers, e.g. 376.6 kJ mol−1 for CH3SiH3.29 Thermal dissociation of C2H6 is dominated by C–C bond breaking, but unimolecular HCl elimination,30,31 with a barrier of 230.1 kJ mol−1, is the dominant reaction for CH3CH2Cl. Could the HCN+HCl channel be more important for the N+CH2Cl reaction than the HCN+H2 channel for N+CH3? By analogy with the N+CH3 reaction, one could envisage the following steps:
| | 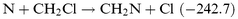 | (R6) |
| | 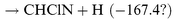 | (R7) |
| |  | (R8) |
| | 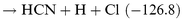 | (R9) |
For
(R7), the enthalpy of reaction is given with a question mark as there is no experimental or theoretical estimate of the enthalpy of formation of CHClN. The C–Cl and C–H bond energies in CH
2Cl are nearly the same.
32 However, the C–Cl bond may be weaker than the C–H bond in the NCH
2Cl adduct. If one assumes that the C–H bond energies in CH
3N and CH
2ClN are similar, the enthalpy of reaction for
(R7) should be closer to that of
(R3)
(−167.4 kJ mol
−1) and
(R7) is certainly less exoergic than
(R6). In our experiments,
(R6) and
(R7) are not directly followed. The HCN and HCl from
(R8) can both be observed by IRCL, if the products are vibrationally excited. In the present work, CH
2ClI was added to a flow reactor in which both H and N atoms were present, and the additional infrared chemiluminescence from the N
+
CH
2Cl reaction was studied in the presence of the H
+
CH
2ClI reaction. The F
+
CH
3X reactions were also used as a precursor for CH
2X radicals. Although HNC is a minor product, it was observed, and the HNC formation channel needs to be added to the N
+
CH
2X mechanism:
| | 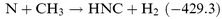 | (R10) |
| |  | (R11) |
During the course of investigating the IRCL from (R2) and (R8), the intense yellow CN(A2Π–X2Σ+) chemiluminescence was clearly evident whenever CH2ClI was added to the flow reactor containing both H and N atoms. The same visible emission was observed on adding CH3Cl to the reactor containing F and N atoms. In fact this CN(A–X) emission was observed for several CH2XI molecules with H and N atoms and for several CH3X molecules with F and N atoms. This CN emission has been commonly observed when halogenated hydrocarbons are added to active nitrogen,33–38 but despite investigations spanning four decades, the excitation-mechanism remains poorly characterized. In the present work, the generation of CH2X
radicals in the presence of N atoms, by the F or H atom primary reactions greatly intensifies the CN emission intensity, and CN formation is also part of the N+CH2X chemistry. The chemiluminescence in the N+CH2X reaction system is discussed and related to the proposed CN excitation mechanisms of the early work.33–38
II. Experimental methods
The IRCL apparatus at Kansas State University has been described previously1–4 and only the pertinent details associated with the addition of N atoms to the flow reactor are given here. The H/F/N atoms were produced in microwave discharges of H2, CF4, and N2 diluted in Ar carrier gas. The H2, N2 and CF4 were taken directly from tanks. Quartz discharge tubes, treated with phosphoric acid, were used for H2 and N2 and an alumina discharge tube was used for CF4. The atoms were generated at the entrance of the flow reactor (4 cm diameter Pyrex tube), and the CH2ClI or the other reagent was added 30 cm downstream via a 4-jet inlet. A few experiments were done with CH4, CH3Cl, CH3Br, CH3I and CH2I2 by metering these reagents as gases
from the reservoirs. The infrared emission was observed through a NaCl window, which was located 2.5 cm from the reagent inlet. The reaction time between CH2ClI and the atoms could be varied from 0.2 to 2 ms by varying the pumping speed with a throttling valve. The reagents, CH2ClI and CH2FI, were introduced to the flow reactor by flowing Ar over the pure liquid kept at 273 K or by flowing the gas from a reservoir containing the liquid and gas in equilibrium. The concentrations of the reagents and the atoms were in the range 1012 to 1013 molecules cm−3. All the experiments were carried out at room temperature at a pressure in the range 0.6–2.0 Torr. The CH2FI was synthesized by the method described in the literature.39 The purity of the CH2FI samples was confirmed by GC-MS to be >90%. The CH2ClI samples were purchased from Aldrich
Chemical Company or from TCI America Inc. (>98% pure) and the purity confirmed by NMR spectra. All the reagents were subjected to several freeze–pump–thaw cycles before use.
The infrared chemiluminescence at 2 cm−1 resolution was observed using a Biorad FTS-60 with a liquid-N2 cooled InSb detector. The HF/DF/HCl/DCl emission spectra were rotationally resolved and the vibrotational distributions were calculated from the peak heights of the P and R branch lines using the Einstein coefficients40 for the given transition and the instrumental response function. The rotational distribution corresponded to 298 K Boltzmann distribution. The relative vibrational population was obtained by averaging all the P and R branch lines from the same vibrational level. The Einstein coefficients for the ν3
(CH stretch) 1→0 transitions in HCN (77 s−1)41 and HNC (385 s−1)42 are much larger than that for HCl (40 s−1),
but the HCN/HNC signals were much weaker than that of HCl. The rotational structure of the HCN/HNC spectra was not resolved, and the area under the band was used to estimate the relative [HCN(v)].
The CN(A→X) emission was recorded at 1.1 Torr pressure from the F+N+CH4/CH3Cl reaction system in a second flow reactor with a reaction time of typically 5 ms. The emission was observed through quartz windows with a 0.5 m monochromator fitted with a cooled S-1 photomultiplier tube. This flow reactor and the spectroscopic detection equipment are described in ref. 43.
III. Results
III.1 Thermochemistry
In order to interpret the IRCL data, accurate thermochemistry is required. If the thermochemistry is unknown, the highest observed HX(v, J) level can be used to set a lower limit on the enthalpy of reaction.44 Experimental enthalpies of formation, ΔfHo, are not available for CH2ClI and CH2FI. However, a theoretical ΔfHo is available for the CH2ClI (13 kJ mol−1)12 calculated at the MP2/3-21++G(2d,2p) level. The ΔfHo for CH2I calculated12 at this level was 218.4 kJ mol−1, in close agreement with the experimental value of 217.6 kJ mol−1. These enthalpies of formation, along with that of Cl (119.62±0.05
kJ mol−1),45 lead to a C–Cl bond energy of 326.4 kJ mol−1 in CH2ClI compared to 351.5±4.2 for CH3Cl.46 Using ΔfHo
(HCl, −92.0±0.5 kJ mol−1),45 the enthalpy of reaction for (R2) is estimated as given below: | |  | (R2) |
The average energy available to the products from a reaction is calculated as:
where Ea is the activation energy for the reaction and Eth is the thermal energy of the reactants. The activation energy for this reaction has not been measured. However, comparison of HCl(v) emission intensities from reaction (R2)
and H+Cl2 suggest that the rate constant for (R2)
(vide infra) is 4 times smaller than for the latter reaction.47,48 The Ea for (R2) is, probably, higher than that (4–8 kJ mol−1) of H+Cl2. We take the Ea to be ≈17 kJ mol−1. The thermal energy of CH2XI at 300 K was calculated to be 12.6 kJ mol−1. Hence, the 〈E〉 for (R2) should be about 133.9 kJ mol−1. This is sufficient to produce HCl(v=4) from (R2), but the highest observed level is v=5. Fig. 1 shows
the HCl(v) IRCL spectrum, highlighting unambiguously the lines from v=5. The additional energy needed for producing v=5, J=0 is 25.1 kJ mol−1. The rotational distribution observed was thermal Boltzmann and HF(v=5, J=0) can be used as the limit. The uncertainty in the theoretical thermochemistry and/or reactive collisions from the high energy tail of the Boltzmann distribution could be the cause of this observation. If HF(v=5) is used to fix the available energy from (R2), then the C–Cl bond energy in CH2ClI will be 301.2 kJ mol−1, 25.1 kJ mol−1 less than the theoretical estimate.12 Inspection of the D(C–Cl)
values from several CH2ClX molecules supports a trend of lower bond energies relative to CH3Cl.46
![The HCl(v) emission spectrum observed from the H + CH2ClI reaction at 0.8 Torr for the reaction time of 0.4 ms. [H] = 2.1 × 1013 and [CH2ClI] = 2.5 × 1013 molecules cm−3. The asterisk indicates the strong lines of the v = 5 → 4 band.](/image/article/2002/CP/b108362g/b108362g-f1.gif) |
| | Fig. 1 The HCl(v) emission spectrum observed from the H+CH2ClI reaction at 0.8 Torr for the reaction time of 0.4 ms. [H]=2.1×1013 and [CH2ClI]=2.5×1013 molecules cm−3. The asterisk indicates the strong lines of the v=5→4 band. | |
The HCl (v=5) emission led us to consider reactions that could take place in the reactor in addition to (R2).
| | 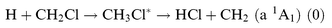 | (R12) |
| |  | (R13) |
The asterisk indicates a chemically activated species. Reaction
(R12) is thermoneutral and it cannot account for HCl(
v) emission. Reaction
(R13) could certainly contribute to HCl(
v) emission. Kinetic simulation was done with the following steps to determine the importance of
(R13):
The
k1 and
k2 are taken from our results (
vide infra) and the value of
k13 is from
ref. 49. After 1 ms, the [CH
2![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif)
CHCl]
(which is the same as [HCl] produced from
(R13) was found to be 10% of the total [HCl]. For the actual reaction time of 0.4 ms, the [CH
2CHCl] was even less at 3% of [HCl]. Hence, the contribution from
(R13) to HCl(
v) emission should be minimal. Moreover, the HCl(
v) distribution from unimolecular elimination reactions declines monotonically with very little population in higher
v levels.
1,2 Hence,
(R12) and
(R13) are ruled out as possible sources of the observed HCl(
v) emission, especially from
v≥
4. Finally, the first-order
dependence of the HCl(
v) intensity on [H] and [CH
2ClI] was verified as proof that
(R2) was the major source for HCl emission, see
Fig. 2. It is concluded that the
ab initio estimate
12 of the C–Cl bond energy in CH
2ClI, 326.4 kJ mol
−1, is an upper limit and it could be as low as 301.3 kJ mol
−1.
![The HCl(v) intensity vs. [H] showing the first-order dependence. The pressure (1.6 Torr), reaction time (0.8 ms) and [CH2ClI]
(4.5 × 1013 molecules cm−3) were kept constant.](/image/article/2002/CP/b108362g/b108362g-f2.gif) |
| | Fig. 2 The HCl(v) intensity vs. [H] showing the first-order dependence. The pressure (1.6 Torr), reaction time (0.8 ms) and [CH2ClI]
(4.5×1013 molecules cm−3) were kept constant. | |
For CH2FI, no theoretical estimate of D(F–CH2I) is available. Hence, the highest HF(v) level observed is used to estimate the lower limit for the C–F bond energy. The HF(v=3, J=0) was used as the limit. With this limit, 〈E〉 is estimated to be ≥138.1 kJ mol−1. The Ea for (R12) was assumed to be 21 kJ mol−1, consistent with the slower reaction rate compared to (R2). This leads to a C–F bond energy of ≤460.2 kJ mol−1 and the enthalpy of reaction is ≤−104.6 kJ mol−1:
| |  | (R14) |
The
D(F–CH
2Cl)
50 and
D(F–CH
3)
51 are ∼460.2 kJ mol
−1 and
halogen substitution does not seem to affect the C–F bond dissociation energy of halofluoromethanes.
III.2 HCl(v) distribution and HCl(v) formation rate constant for H/D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) +CH2ClI
+CH2ClI
The HCl(v) emission could be observed at 0.6 Torr pressure with the maximum pumping speed corresponding to a reaction time of 0.4 ms for [H] and [CH2ClI]≤3×1013 molecules cm−3. Under these conditions,1,2,52,53 there is very little HCl(v) relaxation and the observed distribution is the nascent HCl(v) distribution from the H+CH2ClI reaction. The nascent HCl(v) distribution is P1–P5=25 : 29 : 26 : 13 : 7 obtained as the average of several experiments (Table 1). The HCl(v) distribution was reproducible when experiments were repeated under the same conditions using several different commercial samples of CH2ClI. The
distribution is nearly flat for v=1–3 and it is similar to the HX(v) distribution observed from the H+IX reactions.17–19 For comparison, the HX distributions from H+ICl, NFCl2, CF3O and CF2Cl are included in Table 1. The H+CF2Cl and CF3O reactions give stable molecules before unimolecular HX elimination occurs. In these unimolecular elimination processes, the HCl(v) distribution declines monotonically and 〈fv〉=0.15.1 The H+NFCl2 reaction52 is representative of direct abstraction reactions for which sharply peaked distributions that do not extend to the thermochemical limit
are found. The distributions from H+IX are thought to be the sum of a direct abstraction plus an addition migration step. In this case, the distributions do extend to the thermochemical limit.44
Table 1 HX(v) distribution from various reactions
| Reaction |
Percentage |
〈fv〉
g |
| P1 |
P2 |
P3 |
P4 |
P5 |
|
This work: [H2]=2.0×1013 and [CH2ClI]=4.4×1012 molecules cm−3; P=1.0 Torr, reaction time=0.4 ms. See text for P0.
This work: [H]=6.0×1013 and [CH2FI]=4.2×1013 molecules cm−3. See text for P0.
Ref. 17 The distribution extends to v=7 and the percentage of P6=19; and of P7=14.
Ref. 52.
Ref. 53.
Ref. 2.
Except for H+NFCl2, the 〈fv〉 are based on the surprisal estimates for the amount of P0. Halogen abstraction reactions do not give a linear surprisal and for H+NFCl2, the amount of P0 was assumed to be half that of P1.
|
| H+CH2ClIa |
25 |
29 |
26 |
13 |
7 |
0.48 |
| H+CH2FIb |
76 |
16 |
8 |
— |
— |
0.44 |
| H+IClc |
6 |
12 |
15 |
16 |
18c |
0.61 |
| H+NFCl2d |
20 |
41 |
31 |
8 |
— |
0.31 |
| H+CF3Oe |
34 |
30 |
20 |
10 |
6 |
0.14 |
| H+CF2Clf |
58 |
27 |
10 |
5 |
— |
0.13 |
The IRCL experiments do not give the population of the v=0 level directly. Linear surprisal analysis has been used to estimate the HCl(v=0) populations for the analogous H+ICl reaction.44 The surprisal plot (with model 3 prior which takes all the degrees of freedom for HCl and the other product CH2I) for H+CH2ClI was linear with a correlation coefficient of 0.958. The HCl(v) distribution obtained was P0–P5=1.3 : 24.7 : 31.6 : 24.7 : 10.8 : 6.9 which implies that the P0 is almost negligible. The fraction of available energy going into HCl vibration, 〈fv〉, is calculated to be 0.48. If we assume the P0 to be 1/2 of P1, then 〈fv〉 is 0.44. The available
energy was taken as 167.4 kJ mol−1 in surprisal calculations consistent with the highest HCl(v, J) level observed in the reaction.
The rate constant for the HCl(v) formation channel was estimated by comparing the [HCl(v)] produced from H+CH2ClI with that of H+Cl2 for the same [H] and reaction time. Fig. 3 shows the [HCl(v)]
vs. [CH2ClI] and [Cl2]. The ratio of the slopes of the two lines gives the ratio (0.25) of the rate constants for these two reactions. The rate constant47,48 for H+Cl2 is 2.0×10−11 cm3 molecule−1 s−1. From this, the HCl(v) formation rate constant for (R2) is estimated to be 5±2×10−12
cm3 molecule−1 s−1. The total rate constant for H+CH2ClI has not been measured yet. As noted in the Introduction, the H+CH3I reaction has a rate constant6 of 8.3×10−12 cm3 molecule−1 s−1 and that5 for H+CF3I is 1.4×10−11. The C–I bond in CH2ClI12 is about 12.6 kJ mol−1 weaker than that for CH3I. Hence, the total rate constant for the H+CH2ClI reaction should be ≥2×10−11 cm3 molecule−1 s−1. The HCl(v>0) formation, probably, accounts for ≤25%
of the total reaction.
![The [HCl(v)]
vs. [R] for R = Cl2 and CH2ClI. The [HCl(v)] is the total concentration of [HCl(v)] obtained from the observed intensities corrected for Einstein coefficients and the instrumental response function for all v > 0. From the slopes of the two graphs, the rate constant for the CH2ClI reaction is determined to be one quarter of that for the Cl2 reaction i.e. the rate constant for H + CH2ClI → HCl(v > 0) + CH2I is 5 ± 2 × 10−12 cm3 molecule−1 s−1.](/image/article/2002/CP/b108362g/b108362g-f3.gif) |
| | Fig. 3 The [HCl(v)]
vs. [R] for R=Cl2 and CH2ClI. The [HCl(v)] is the total concentration of [HCl(v)] obtained from the observed intensities corrected for Einstein coefficients and the instrumental response function for all v>0. From the slopes of the two graphs, the rate constant for the CH2ClI reaction is determined to be one quarter of that for the Cl2 reaction i.e. the rate constant for H+CH2ClI→HCl(v>0)+CH2I is 5±2×10−12 cm3 molecule−1 s−1. | |
The D+CH2ClI reaction gave both HCl(v) and DCl(v) emission, see Fig. 4. The DCl(v) emission was weak due to the much smaller (≈1/4 of that of HCl) Einstein coefficient. In order to get a better signal to noise ratio, these experiments were done at longer observation time (0.8 ms) and higher concentrations; both [D2] and [CH2ClI] were 5–10×1013 molecules cm−3. Under these conditions, the HCl(v) distribution from H+CH2ClI was somewhat relaxed with a distribution of P1–P4=55 ∶ 31 ∶ 11 ∶ 3 compared to the nascent distribution reported in Table 1. From the D+CH2ClI
reaction, the distributions were 57 ∶ 31 ∶ 10 ∶ 2 for HCl(v) and 36 ∶ 33 ∶ 24 ∶ 7 for DCl(v). The ratio of DCl(v≥1)/HCl(v≥1) concentrations observed in the experiments was about 1.1. At larger reagent concentration, this ratio was reduced to 0.7. This may be the result of more efficient DCl(v) relaxation compared to HCl(v) relaxation, with more DCl(v=0) being present. The HCl formation from D+CH2ClI with a similar distribution to that for H+CH2ClI, shows that extensive H/D exchange occurs in the DICH2Cl adduct. The other possibility could be elimination from both the iodine and carbon atom sites. But, then one would expect more different HCl(v) and DCl(v)
distributions.
![The IRCL spectrum from the D + CH2ClI reaction with (upper) and without (lower) N atoms. The reactor pressure was 1.6 Torr for the lower spectrum and 1.7 Torr for the upper spectrum. Both [H] and [CH2ClI] ≈ 5 × 1013 molecules cm−3. Reaction time was 0.8 ms.](/image/article/2002/CP/b108362g/b108362g-f4.gif) |
| | Fig. 4 The IRCL spectrum from the D+CH2ClI reaction with (upper) and without (lower) N atoms. The reactor pressure was 1.6 Torr for the lower spectrum and 1.7 Torr for the upper spectrum. Both [H] and [CH2ClI]≈5×1013 molecules cm−3. Reaction time was 0.8 ms. | |
III.3 HF(v) distribution from H+CH2FI
The amount of CH2FI sample was very limited and only a few experiments could be done. The HF(v) emission from the H+CH2FI reaction could be seen only with reduced pumping speed, thus the reaction time was 0.7 ms for [H] and [CH2FI] in the range 5–10×1013 molecules cm−3. The HF(v) distribution observed was P1–P3= 77 ∶ 15 ∶ 8. In general, HF(v) relaxation is slower than HCl(v) relaxation due to the larger energy spacings. Hence, vibrational relaxation is unlikely to be the main reason for the much lower HF(v) excitation than HCl(v) excitation from H+CH2ClI. Comparison of [HF(v)] with [HCl(v)] from
H+Cl2 implies that the HF(v) formation rate is about 20 times slower than the H+Cl2 reaction. This indicates that the adduct formation followed by HF formation is a small part of the total H+CH2FI reaction. A casual look at HF(v>0) distribution suggests similarity to that of addition–elimination reactions,1,2 except for the fact that the energy available to the HF(v) products is much smaller than the HX(v) elimination reactions reported in ref. 1 and 2. This is evident from the results of surprisal analysis (which had a correlation coefficient slightly smaller than for (R2) at 0.943). The HF(v) distribution obtained was P0–P3=0.7
: 75.4 : 15.9 : 7.9 and the 〈fv〉 was 0.44. Although, this estimate is an upper limit, the 〈fv〉 seems to be comparable to that for the H+CH2ClI reaction.
The D+CH2FI reaction gave both HF and DF emission. The HF(v) distribution appeared to indicate a smaller 〈fv〉 with P1–P3=87 ∶ 13 ∶ 0, compared to that of H+CH2FI discussed above. The DF(v) distribution was 71 ∶ 21 ∶ 8. The [HF(v)] was about 2 times smaller than the [DF(v)]. As already pointed out, the CH2FI sample was very limited and more experiments to get the nascent distribution from this reaction could not be performed. However, these results are significant in establishing H/D exchange unambiguously, as found for the D+CH2ClI reaction.
III.4 N+CH2X reactions: HX+HCN channel
The changes in HX(v) distribution and intensity were monitored on addition of N atoms to the H+CH2XI and F+CH3X reaction systems. In order to demonstrate the effect of N atoms and N2, reference spectra were taken at the same pressure without a N2 flow. In the second step spectra were obtained with added N2 and finally the N/N2 experiment was done; the reaction time was 0.8 ms and the pressure was 1.7 Torr. Results for the H+CH2ClI are shown in Table 2
(entries 1–3). The HCl(v) distribution observed under these conditions (without added N2) was P1–P4=37 ∶ 32 ∶ 22 ∶ 9, which shows the effects of some relaxation. When N2 was added with the microwave discharge
off, there was very little change in the HCl(v) distribution or the total emission intensity. When the N2 discharge was turned on, the HCl(v) intensity nearly doubled for the most favorable experimental conditions and the distribution became P1–P4=45 ∶ 28 ∶ 18 ∶ 9. In addition, the HCN (001→000) emission could be observed. This large increase in the IHCl is not necessarily a contradiction because the H+CH2ClI reaction probably gives more CH2Cl (+HI) radicals than CH2I (+HCl) radicals in the primary step. The increase in HCl(v) intensity and the observation of concomitant HCN emission in the presence of N atoms are convincing evidence for (R8). The overall HCl(v)
distribution showed very little change, on addition of N atoms. It is likely that the HCl(v) distribution from the N+CH2Cl reaction is similar to that of the H+CH2ClI reaction. It should be noted that the H+CH2Cl (k≈2–4×10−11 cm3 molecule−1 s−1)54 and N+CH2Cl (k≈8×10−11 cm3 molecule−1 s−1)21 are in competition. However, as already pointed out, the formation of HCl from H+CH2Cl is thermoneutral and cannot be observed.
Table 2 HCl(v)/DCl(v) distribution and total observed emission intensity from H/D+CH2ClI with/without N2/N in the reactora
| No |
Atom/XCl(v) |
Percentage |
ΣHCl(v) |
| P1 |
P2 |
P3 |
P4 |
|
Experiments 1–3 had [H]=6×1013, [CH2ClI]=1.5×1013 molecules cm−3 and [N2]=2.3×1015. Pressure=1.7 Torr and the reaction time was 0.8 ms. For experiments 4–9, high [CH2ClI] was needed, 1.9×1014 molecules cm−3, to obtain a good signal to noise ratio for the DCl(v) emission. At these concentrations, the HX(v) distribution showed some relaxation compared to that given in Table 1 and emission from v=5 was absent due to relaxation. (see text).
|
| 1 |
H/HCl |
36.6 |
32.3 |
22.2 |
8.9 |
0.057 |
| 2 |
H/HCl+N2 |
38.0 |
33.5 |
20.3 |
8.2 |
0.063 |
| 3 |
H/HCl+N |
45.4 |
27.1 |
17.7 |
8.0 |
0.093 |
| 4 |
D/DCl |
35.6 |
32.8 |
24.3 |
7.3 |
0.092 |
| 5 |
D/DCl+N2 |
38.3 |
29.2 |
24.8 |
7.7 |
0.082 |
| 6 |
D/DCl+N |
57.1 |
27.7 |
8.4 |
6.7 |
0.115 |
| 7 |
D/HCl |
56.9 |
31.0 |
10.1 |
2.0 |
0.135 |
| 8 |
D/HCl+N2 |
54.0 |
33.7 |
10.2 |
2.1 |
0.124 |
| 9 |
D/HCl+N |
56.8 |
30.5 |
10.7 |
2.0 |
0.172 |
Similar observations were made with the D+N+CH2ClI system. The IRCL spectrum showing HCl, DCl and HCN emission from this system is shown in Fig. 4. When no N atoms are present in the reactor, only HCl and DCl emission is observed. With added N atoms, HCl and DCl intensities both increase and weak HCN emission is observed. Entries 4–9 in Table 2 summarize our observation. The HCl(v) distribution did not change significantly with the presence of N2 or N. However, the DCl(v) distribution showed a detectable difference in the presence of N atoms but not with N2 alone. From the above observations, it appears that the DCl(v) and HCl(v) distributions from the N+CH2Cl/CHDCl may be peaking at lower v. For both HCl and DCl, the change
in distribution is rather subtle and it is not possible to make any more meaningful quantitative interpretation.
The addition of N atoms to the H/D+CH2FI system led to an increase in HF(v) and DF(v) emission intensity along with weak HCN(ν3) emission. Separate experiments were done for the H+CH2I2+N and H+CH3I+N reactions. All experiments gave weak HCN(ν3) emission and even weaker HNC emission, as shown in Fig. 5. Thus, the N+CH2X reactions do have some general nature for X=H, Cl, F and I. The HCN emission observed from these reactions (for X=Cl and F) mainly consisted of the (001)→(000) transition with little evidence for any bending or CN stretch excitation. However, the H+CH2I2+N
reaction system did generate additional vibrational excitation in HCN, see Fig. 5. For the H+N+CH2ClI reaction system, the [HCN(v)] was estimated from the area under the curve, correcting for the Einstein coefficient and instrument response function. The [HCN(v3=1)] was 10% of the increase in [HCl(v)]. If the increase in HCl(v) and HCN(v) are due to the same reaction (R8), then one would expect equal amounts of HCl(v) and HCN(v). It is likely that most of the HCN product is produced in the vibrational ground state. However, this is just a speculation, since our experiment cannot directly determine the ground state population for HCN or HCl.
![The HCN(v) and HNC(v) infrared emission spectra observed from H + N + CH2I2 reactions. The pressure was 1.8 Torr and the reaction time was 0.8 ms. [H] = 6 × 1013. Both HCN and HNC emission are thought to result from the N + CH2I reaction.](/image/article/2002/CP/b108362g/b108362g-f5.gif) |
| | Fig. 5 The HCN(v) and HNC(v) infrared emission spectra observed from H+N+CH2I2 reactions. The pressure was 1.8 Torr and the reaction time was 0.8 ms. [H]=6×1013. Both HCN and HNC emission are thought to result from the N+CH2I reaction. | |
Experiments also were done with N+CH3, N+CH2Cl and N+CH2Br using the F+CH4, CH3Cl and CH3Br as primary reactions. Unfortunately the strong HF(v,J) emission overlaps any weak HCN emission. Under favorable conditions, the HCN emission could be clearly observed underneath the HF(v,J) emission as a shift in the base line. We attempted to observe an increase in HCl emission from F+N+CH3Cl compared to F+CH3Cl. The latter reaction system gives both HF from the F+CH3Cl primary reaction and HCl from the F+CH2Cl secondary reaction.1 The experiments were inconclusive because of several problems. We were
unable to obtain [F] above 3–4×1012 molecule cm−3. Thus, the CH2Cl was lower than in the H atom experiments. In addition, the discharge products from CF4 and N2 interacted as evidenced from a chemiluminescence in the pre-reactor. Moreover, both HF(v) and HCl(v) intensities increased when N atoms were added. The additional HF is probably due to the production of more F atoms from the N+CFn reactions. However, the CN chemiluminescence seemed to be of normal intensity, which implies that the (N) was normal. Observation of both HCN and CN emission (next section) provides evidence for the N+CH2X reaction in these systems.
III.5 N+CH2X reactions: CN emission
Both H+N+CH2XI and F+N+CH3X (X=H, F, Cl, Br and I) reaction systems gave strong CN (A→X) emission that was readily visible in a slightly darkened room. An emission spectrum was recorded for the F+CH3Cl/CH4+N reaction systems, using the second flow reactor. The vibrational distribution resembles the P2 distribution characterized by Bayes36 but with somewhat higher relative populations in the low v levels. This emission is the peach-colored flame observed when halogenated hydrocarbons are added to active nitrogen.33,36 The H+CH2XI and F+CH3X systems both led to the CN emission, clearly indicating that
the F/H atoms do not have any role except to produce the CH2X radicals. This also rules out the CFn species, which may be present in the CF4 discharge, as the only sources of CN. At our flow velocities and concentrations, the N/N2+CH3X or CH2IX reactions alone gave no emission that was visible to the eye or the IR detector. Benson and coworkers55 have studied the N+CHCl3 reaction in a very low pressure reactor for longer reaction time (a few seconds) and reported a rate constant of 1×10−16 cm3 molecule−1 s−1 for the primary step. This is consistent with our observation of no CN chemiluminescence in the absence of any added F or H atoms. In our reactors, the CN emission began at the reagent inlet as soon as the F or H atom discharge
sources were activated and the onset of the chemiluminescence seems to depend on the mixing rate of the reagent into the gas flow. In the absence of CH3X, the discharge products from N2 and CF4 reacted, generating a weak unidentified emission, in the pre-reactor section of the flow reactor for high N2 and CF4 flows. However, this unidentified reaction must be relatively slow, and the emission extended over the whole length of the reactor. The addition of CH3X reagents to the N/F/CFx flow gave much stronger emission, which obscured this weak emission. Another observation was the build-up of (CN)x polymer on the flow reactor beyond the reagent inlet, as CN radical sources are prone to do.26,27 This suggests that the CN concentration is not negligible, and that the N+CH2X reaction system must
include CN-generating steps.
The emission spectra and measurements of the dependence of the CN emission intensity were taken in the reactor that permitted the [N] to be measured by titration with NO and the [F] to be measured by titration with CF3I. The various emission intensities were measured with a 0.5 m monochromator fitted with a cooled S-1 photomultiplier tube. The experiments were done with CH3Cl and CH4 concentrations of ∼3×1013 molecules cm−3 with [F]=3×1012 and [N]=0.5–5×1012 atoms cm−3. For a fixed [N] concentration, the CN emission intensity increased linearly with [F] for fixed [CH3Cl] and with [CH3Cl] for fixed [F] providing that the variable concentration was not
so large as to deplete the reagent with the fixed concentration. An experiment to test the order dependence for [N] was done with [CH3Cl]=4×1013 and [F]=2×1012 molecule cm−3. The results, shown in Fig. 6, demonstrate apparent first-order kinetics for [N]=1–5×1012 atoms cm−3.
![The CN(A → X) emission intensity from the F + CH3Cl + N reaction system as a function of [N]. The [N] was determined by titration with NO by observation of the N2(B-A) emission intensity to determine the end point. [F] = 2 × 1012 and [CH3Cl] = 3 × 1013 molecules cm−3; the reaction time was 5 ms.](/image/article/2002/CP/b108362g/b108362g-f6.gif) |
| | Fig. 6 The CN(A→X) emission intensity from the F+CH3Cl+N reaction system as a function of [N]. The [N] was determined by titration with NO by observation of the N2(B-A) emission intensity to determine the end point. [F]=2×1012 and [CH3Cl]=3×1013 molecules cm−3; the reaction time was 5 ms. | |
IV. Discussion
IV.1 The H+CH2ClI and CH2FI reactions
The H+CH2ClI reaction at room temperature has several surprising aspects. Foremost was the HCl(v>0) product channel with a rate constant of 5±2×10−12 cm3 molecule−1 s−1 at 300 K, which is much larger than that for H+CH3Cl or CH2Cl2. A second surprise was the observation of both HCl and DCl formation from D+CH2ClI. Finally the HCl(v) distribution was rather flat with strong emission from HCl(v≤4) and weak emission from v=5, which indicates a low D(Cl–CH2I) as already discussed. All of these aspects are consistent only with addition of the H atom to the iodine atom with a low activation energy followed by
partial exchange of the H atoms between the iodine and carbon atom with subsequent formation of HCl. The mechanism may be somewhat similar to the H+IX reaction systems, which have a direct interaction channel giving HX, plus an indirect channel involving addition to iodine atom followed by H-atom migration to the X atom.17–19 Attack at the iodine atom serves as a way of circumventing the energy barrier for direct abstraction. The HCl(v) distribution gives 〈fv(HCl)〉=0.48, which is much larger than most 3- or 4-centred unimolecular elimination reactions. The linear vibrational surprisal and the large 〈fv〉 are additional evidence for a different mechanism (i.e. neither direct abstraction nor addition–elimination) and the term addition–migration reaction may be
a better description. The Cl and O atom reactions with CH2ClI, F with CH2BrCl and Cl with CH3I also involve an addition pathway. Since iodine forms stable molecules with 10 or 12 electrons around the central iodine atom, free radical addition to give a 9-electron adduct of iodine should not be so surprising. In fact, ab initio calculations for the adducts with Cl atoms do find bent C–I–Cl structures of CH3I and CH2ClI with weak I–Cl bonds (55 kJ mol−1).12,15 The H atom exchange pathway and the HCl or HF formation mechanisms are less easy to visualize. The trajectory calculations for H+ICl may provide some clues.19 After the H atom bonds to the I atom, the H makes many large excursions while the heavy Cl atom begins to depart. In some cases the H atom has an encounter, either repulsive
or attractive, with the Cl atom. For attractive encounters, the H atom is passed to the Cl atom. If the ∠HIC is not linear, as theoretically found for ∠ClIC, large amplitude motions can bring the H atom closer to the Cl and C atoms. For the latter, a synchronous motion of one of the H bound to C toward the I atom lead to exchange of H atom positions i.e., the H atoms originally bound to I and C interchange positions by a rearrangement pathway. Recently, Holmes and coworkers have identified such an unusual 1,2-FCl rearrangement pathway in CF2ClCF2CH3, which facilitates 4-centered HCl elimination through CF3CFClCH3.56
Is the HI formation, (R1), process always direct or can it be an exit channel from the adduct? Certainly the O atom addition mechanism seems to be fairly general for alkyl iodides.13,14 The HCl(v) and DCl(v) intensities reported in Table 2 offer some important clues. Intensities of both HCl(v) and DCl(v) increase on N atom addition to the D+CH2ClI reaction system. If DI formation occurs through direct abstraction only, then one would expect the HCl(v) intensity alone to increase by the contribution from N+CH2Cl, with no change in DCl(v). The fact that both increase implies that H/D exchange occurs before the HI/DI formation and that both CH2Cl and CHDCl radicals are produced. The
DCl(v)
(from N+CHDCl) emission was weak and a more quantitative interpretation is not possible.
IV.2 The N+CH2X reaction (X=H, Cl, F, I)
The primary interaction between N(4S) atom and CH2X radicals is association to give the CH2XN triplet ground-state nitrene radical. The nitrene radical has enough energy for dissociation and (R6) or (R7) is the most important product channel. The CHXN radical probably lacks sufficient energy to overcome the barrier for loss of a second H atom,57–59 but dissociation of a carbon–halogen bond might be possible. The (R4)
(or (R8)) step requires intersystem crossing to the singlet nitrene potential surface,20,57 and this seems to be the rate limiting step. Our IRCL data confirm that (R8)) does occur in competition with (R6)
(or (R7)) for several CH2X type radicals. The HCl(v) distribution appears similar to that of the H+CH2ClI reaction. However, the HCl formation mechanism is not established and both internal conversion and structural isomerization of NCH2Cl may occur prior to HCl elimination. The IRCL results do show that the vibrational energy released to HCl and HCN is a small part of the available energy from (R8). The HCN emission was always weak, but there was some indication that this emission was stronger from molecules with weaker C–X bonds. The singlet methyl nitrene radical has the possibility for isomerization to CH2NH or even CHNH2 and, according to the calculations, 1,1-elimination of H2 is preferred over 1,2-elimination.57 Thus, some HNC formation could be expected,
as we observed. Our data, unfortunately, cannot be placed on a quantitative basis and we cannot assign branching ratios to dissociation of triplet CH2XN vs. the elimination step giving HX and HCN (or HNC). The IRCL spectra do show that HCN and HNC have little vibrational excitation.
Secondary reactions must be responsible for the CN chemiluminescence because N+CH3 does not provide sufficient energy to give CN(A)+HX+H. Since the radical species from the primary steps seem reasonably well established and since the thermochemistry can be estimated, a discussion of the mechanism responsible for the chemiluminescence in the N+CH2X reaction seems worthwhile. Since the CN(X,v) and CN(A,v) states are rapidly interconverted60–62 by collisions with Ar, it may not really matter whether high vibrational levels of CN(X) or CN(A) are initially formed. Therefore, we will just search for reactions with sufficient exoergicity to give CN(A, v′=8). As a consequence of early work on the subject33–38
several excitation steps have been discussed. These include: (i) the N(4S)+CX(X2A) reaction, (ii) excitation by an electronic energy-transfer step, (iii) CN acting as a third body in atomic (N or O) recombination, (iv) more complex CN-forming reactions, such as N+NCN. Dorthe and coworkers63 have isolated the N+CCl reactions and confirmed that (i) does give CN(A) with the P2 type vibrational distribution, as suggested by Thrush and Setser.34,64 The original argument64 in support of this type of reaction was based on the fact that the CN(A) potential lies below the CN(X) potential at long distance. However, these reactions probably proceed by addition of N to the radical
to give a stable triplet adduct that subsequently dissociates,65 rather than by direct X atom abstraction from an attacking N atom. Thus the CN bond is probably formed before the X atom leaves. Except for the N+C+M reaction, the three-body recombination mechanisms can be discounted because nowadays it is established that the two-body reactions of O and N atoms with CN are very fast.37 Benson and coworkers55 have suggested the following sequence to explain the CN emission from the N+CHCl3 reaction system:
| | 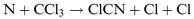 | (R15) |
| |  | (R16) |
| | 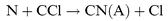 | (R17) |
The N
+
ClCN reaction is very slow in the absence of ‘active
nitrogen’,
34,64 and the second step is doubtful. In our systems, we do not see how CCl(X
2Π) or CH(X
2Π) radicals can be generated, and the electronic transfer steps also do not seem feasible. We believe that the N atoms must react with the product (CH
2N or CHXN) from the N
+
CH
2X reaction. Although easily visible, the CN chemiluminescence is probably associated with a minor channel of the N
+
CHXN chemistry.
We suggest the following sequence of reactions, with the thermochemistry based on X=H and ΔfHo
(CH2N)66=238.5 and ΔfHo
(NCN)67=456.1 kJ mol−1.
| |  | (R18a) |
| |  | (R18b) |
| |  | (R18c) |
| |  | (R19) |
The formation of NCN would be in competition with several other possible product channels as given in
(R18). Moreover, it may occur through a complex mechanism involving more than one step. Steif and coworkers
23
have identified
(R18a) as a dominant channel for X
=
H. A direct route from CHXN to CN would be more desirable; however, formation of CN from N
+
CH
2N seems unlikely. The isomerization of CH
2N to HCNH or CNH
2 have calculated barriers
57 of 188.3 and 288.7 kJ mol
−1 and they are unlikely candidates. Since the CH
2N and NCN radicals are formed and removed by steps involving N atoms, the CN concentration will be approximately first order in [N], in accord with the experimental observations. It is worth noting that McFadden and coworkers
68 observed CN formation when monitoring the products from the N
+
CF
3 reaction by mass spectrometric detection. Most spectroscopic studies of NCN radicals have used the N
2/CF
4/He microwave
discharge as the source for NCN,
69–71 and formation of NCN in our reaction systems and that of McFadden and coworkers
68 could be expected. The mechanism proposed above can explain the very general finding of CN formation from the interaction of a variety of halomethyl radicals with N atoms.
V. Conclusions
Infrared chemiluminescence in a fast flow reactor has been used to identify HCl(v>0) formation from the H+CH2ClI reaction with a rate constant of 5±2×10−12 cm3 molecule−1 s−1 at room temperature. The D+CH2ClI reaction gives both HCl and DCl products, which strongly suggests a mechanism in which H/D exchange occurs in an adduct. The HCl(v) distribution has a linear surprisal with 〈fv〉=0.48, which suggests an analogy to the dynamics for the addition–migration channel of the H+ICl reaction. From the highest observed HCl(v=5) level, D(Cl–CH2I) is estimated to be ≤301.2 kJ mol−1 compared
to a theoretical estimate of 326.4 kJ mol−1. The H+CH2FI reaction is similar to the H+CH2ClI and the highest observed HF(v=3) level from the H+CH2FI reaction implies that the C–F bond energy is ≤460.2 kJ mol−1. The lower D(C–Cl), relative to that of CH3Cl, is consistent with the bond energies of other CH2XCl molecules. In contrast, multiple halogen substitution does not seem to affect the D(C–F) values very much. The I atom in the CH2XI molecules provides a reaction pathway to access the Cl or F atoms with a lower energy barrier compared to direct X abstraction. The H/D exchange was identified by the inference of CH2Cl and CHDCl product as well, which suggests that the HI formation channel also arises via
the adduct.
The addition of N atoms to the reactor containing CH2Cl radicals gave additional HCl(v) emission plus weak HCN and HNC emission. These observations identify a singlet exit channel from the N+CH2Cl reaction, although the spin-conserving triplet channel giving Cl+CH2N products is probably the dominant one. Fast secondary reactions starting with N+CH2N reaction must lead to CN formation, in part, as identified by the CN(A) chemiluminescence from several F/CH3X/N and H/CH2XI/N reaction systems. One possibility for the CN(A) forming step is N+NCN. However, the products from the N+CH2N reaction need to be identified before this possibility could be confirmed.
Acknowledgements
RV acknowledges the support of the National Science Foundation under a REU grant CHE-9732103 to KSU. EA thanks the Director, IISc and IISc-ISRO Space Technology Cell for partial support. We thank G. C. Manke II for communicating unpublished results on H+Cl2 rate constant measurements.
References
- E. Arunan, S. J. Wategaonkar and D. W. Setser, J. Phys. Chem., 1991, 95, 1539 CrossRef CAS.
- E. Arunan, R. Rengarajan and D. W. Setser, Can J. Chem., 1994, 72, 568 CAS.
- A. Srivatsava, E. Arunan, G. Manke, D. W. Setser and R. Sumathi, J. Phys. Chem. A, 1998, 102, 6412 CrossRef CAS.
- N. I. Butkovskaya and D. W. Setser, J. Chem. Phys., 1996, 105, 5064 CrossRef.
- R. A. Morris, K. Donohue and D. L. McFadden, J. Phys. Chem., 1989, 93, 1358 CrossRef CAS.
- P. Marshall, A. Misra and R. J. Berry, Chem. Phys. Lett., 1997, 265, 48 CrossRef CAS.
- J. Yuan, L. Wells and P. Marshall, J. Phys. Chem. A, 1997, 101, 3542 CrossRef CAS.
- R. Vogt, R. Sander, R. Von Glasow and P. J. Crutzen, J. Atmos. Chem., 1999, 32, 375 CrossRef CAS.
- O. V. Rattigan, D.
E. Shallcross and R. A. Cox, J. Chem. Soc., Faraday Trans., 1997, 93, 2839 RSC.
- C. M. Roehl, J. B. Burkholder, G. K. Moortgat, A. R. Ravishankara and P. J. Crutzen, J. Geophys. Res.[Atmos.], 1997, 102, 12819 Search PubMed.
- M. Bilde, J. Sehested, O. J. Nielsen, T.
J. Wallington, R. J. Meagher, M. E. McIntosh, C. A. Piety, J. M. Nicovich and P. H. Wine, J. Phys. Chem. A, 1997, 101, 8035 CrossRef CAS.
- K. G. Kambanis, D. Y. Argyris, Y. G. Lazarou and P. Papagiannakopoulos, J. Phys. Chem. A, 1999, 103, 3210 CrossRef CAS
(The experimental value for the CH2I enthalpy of formation
has been quoted in this paper from W. B. DeMore, S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo, C. J. Howard, A. R. Ravishankara, C. E. Kolb and M. J. Molina, Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling, JPL Publication 97-4, 1997.
- X. Gao, J. Essex-Lopresti, S. Munro, M. P. Hall, D. J. Smith and R. Grice, J. Phys. Chem. A, 1998, 102, 1912 CrossRef CAS.
- R. A. Loomis, J. J. Klaassen, J. Linder, P. G. Christopher and S. R. Leone, J. Chem. Phys., 1997, 106, 3934 CrossRef CAS.
- Y. V. Ayhens, J. M. Nicovich, M. L. McKee and P. H. Wine, J. Phys. Chem. A, 1997, 101, 9382 CrossRef CAS.
- M. Bilde, J. Sehested, O. J. Nielsen and T. J. Wallington, J. Phys. Chem. A, 1997, 101, 5477 CrossRef CAS.
- K. Tamagake and D. W. Setser, J. Phys. Chem., 1979, 83, 1000 CAS.
- D. Brandt and J. C. Polanyi, Chem. Phys., 1980, 45, 65 CrossRef CAS.
- J. C. Polanyi, J. L. Schreiber and W. J. Skrlac, Faraday Discuss Chem. Soc., 1979, 67, 66 RSC.
- R. G. Sadygov and D. R. Yarkony, J. Chem. Phys., 1997, 107, 4994 CrossRef CAS.
- L. J. Steif, G. Marston, D. F. Nava, W. A. Payne and F. L. Nesbitt, Chem. Phys. Lett., 1988, 147, 570 CrossRef.
- G. Marston, F. L. Nesbitt, D.
F. Nava, W. A. Payne and L. J. Steif, J. Phys. Chem., 1989, 93, 5769 CrossRef CAS.
- G. Marston, F. L. Nesbitt and L. J. Stief, J. Chem. Phys., 1989, 91, 3483 CrossRef CAS.
-
D. F. Davidson and R. K. Hanson, in Proceedings of the 23rd Symposium (International) on Combustion, The Combustion Institute, Pittsburgh, PA, 1990,
p. 267 Search PubMed.
- Y. L. Yung, M. Allen and J. P. Pinto, Astrophys. J. Suppl., 1984, 55, 465 CrossRef CAS.
- E. Arunan and Setser, J. Phys. Chem., 1991, 95, 4190 CrossRef CAS.
- E. Arunan, G. Manke II and D. W. Setser, Chem. Phys. Lett., 1993, 207, 81 CrossRef CAS.
-
(a) L. R. Copeland, F. Mohammad, M. Zahedi, D. H. Volman and W. M. Jackson, J. Chem. Phys., 1992, 96, 5897 CrossRef;
(b) L. R. Copeland, F. Mohammad, M. Zahedi, D. H. Volman and W. M. Jackson, J. Chem. Phys., 1992, 97, 3878 CrossRef CAS.
- M. S. Gordon and T. N. Truong, Chem. Phys. Lett., 1987, 142, 110 CrossRef CAS.
- W. G. Clark, D. W. Setser and K. Dees, J. Am. Chem. Soc., 1971, 93, 5328 CrossRef CAS.
- K. Dees and D. W. Setser, J. Chem. Phys., 1968, 49, 1193 CrossRef CAS.
- C. F. Rodriquez, D. K. Bohme and A. C. Hopkinson, J. Phys. Chem., 1996, 100, 2942 CrossRef CAS and references cited therein.
- T. Iwai, M. I. Savadatti and H. P. Broida, J. Chem. Phys., 1967, 47, 3861 CrossRef CAS.
- D. W. Setser and B. A. Thrush, Proc. R. Soc. London, Ser A., 1965, 288, 256 Search PubMed and 275.
- J. C. Boden and B. A. Thrush, Proc. R. Soc. London, Ser A., 1968, 305, 93 Search PubMed and 107.
- K. D. Bayes, Can. J. Chem., 1961, 39, 1074 CAS.
- M. R. Gorbal and M. I. Savadatti, Chem. Rev., 1982, 82, 527 CrossRef CAS.
- C. A. Arrington Jr., O. O. Bernardini and G. A. Kistiakowsky, Proc. R. Soc. London, Ser A., 1969, 310, 161 Search PubMed.
- R. N. Haszeldine, J. Chem. Soc., 1952, 4259 RSC.
- E. Arunan, D. W. Setser and J. F. Ogilvie, J. Chem. Phys., 1992, 97, 1734 CrossRef CAS.
- P. Botschwina, Chem. Phys., 1983, 81, 73 CrossRef CAS.
- J. D. Rogers and J. J. Hillman, J. Chem. Phys., 1982, 77, 3615 CrossRef CAS.
- K. B. Hewett, G. Manke II, D. W. Setser and G. Breewood, J. Phys. Chem. A, 2000, 104, 539 CrossRef CAS.
-
B. E. Holmes and D. W. Setser, in Physical Chemistry of Fast Reactions, ed. I. W. M. Smith, Plenum Press, New
York, 1980. Search PubMed.
- M. W. Chase, Jr., C. A. Davies, J. R. Downey Jr., D. J. Frurip, R. A. McDonald and A. N. Syverud, J. Phys. Chem. Ref. Data., 1985, 14 , (Suppl. No 1).
- A. K. Chandra and T. Uchimaru, J. Phys. Chem. A, 2000, 104, 9244 CrossRef CAS.
- D. Kita and D. H. Stedman, J. Chem. Soc., Faraday Trans. 2, 1982, 78, 1249 RSC.
- P. P. Bemand and M. A. A. Clyne, J. Chem. Soc., Faraday Trans. 2, 1977, 73, 394 RSC . (A recent measurement by O.Dobis and S.W. Benson suggests a rate constant that is 1/2 of the value given by refs. 47 and 48. J. Phys. Chem. A, 2000, 104, 777. However, unpublished results by G. C. Manke II and M. C. Heaven support the earlier results.).
- P. B. Roussel, P. D. Lightfoot, F. Caralp, V. Catoire, R. Lesclaux and W. Forst, J. Chem. Soc., Faraday Trans., 1991, 87, 2367 RSC.
- A. A. Zavitsos, J. Phys. Chem., 1987, 91, 5573 CrossRef.
- D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493 CrossRef CAS.
- E. Arunan, C. P. Liu, D. W. Setser, J. V. Gilbert and R. D. Coombe, J. Phys. Chem., 1994, 98, 494 CrossRef CAS.
- R. Rengarajan, D. W. Setser and D. D. DesMarteau, J. Phys. Chem., 1994, 98, 10568 CAS.
- Atom–radical reactions are usually fast. For example the H+CF3 reaction has a rate constant of 8.9×10−11 cm3 molecule−1 s−1:
K. R. Ryan and I. C. Plumb, Plasma Chem. Plasma Process., 1984, 1, 141.
- S. C. Jeoung, K. Y. Choo and S. W. Benson, J. Phys. Chem., 1991, 95, 7282 CrossRef CAS.
- M. O. Burgin, G. L. Heard, J. M. Martell and B. E. Holmes, J. Phys. Chem. A, 2001, 105, 1615 CrossRef CAS.
- M. T. Nguyen, D. Sengupta and T.-K. Ha, J. Phys. Chem., 1996, 100, 6499 CrossRef.
- B. S. Jursic, J. Chem. Soc., Faraday Trans., 1997, 93, 2355 RSC.
- P. Felder, J. A. Harrison and J. R. Huber, J. Phys. Chem., 1991, 95, 1945 CrossRef CAS.
-
(a) G. Jihua, A. Ali and P. J. Dajdigian, J. Chem. Phys., 1986, 85, 7098 CrossRef CAS;
(b) G. Jihua, A. Ali and P. J. Dajdigian, J. Chem. Phys., 1987, 87, 2045 CrossRef.
- N. Furio, A. Ali and P. J. Dajdigian, J. Chem. Phys., 1986, 85, 3860 CrossRef CAS.
- M. de Moor, Ch. Ottinger, A. F. Vilesov and D. D. Xu, J. Chem. Phys., 1994, 101, 9506 CrossRef CAS.
- N. Daugey, A. Bergeat, J.-C. Loison, A. Schuck, P. Caubet and G. Dorthe, Chem. Phys. Lett., 2000, 324, 1 CrossRef CAS.
- D. W. Setser and B. Thrush, Nature, 1963, 200, 864 CAS.
- M. T. Rayez, Ph. Halvick, J. C. Rayez, Ph. Millie’ and B. Levy, Chem. Phys., 1994, 188, 161 CrossRef CAS.
-
(a) R. Sumathi and M. T. Nguyen, J. Phys. Chem. A, 1998, 102, 8013 CrossRef CAS;
(b) D. C. Cowels, M. J. Travers, J. L. Freuh and G. B. Ellison, J. Chem. Phys., 1991, 94, 3517 CrossRef;
(c) D. C. Cowels, M. J. Travers, J. L. Freuh and G. B. Ellison, J. Chem. Phys., 1991, 95, 3864 CrossRef report 213±21 kJ mol−1.
- L. V. Moskaleva and M. C. Lin, J. Phys. Chem. A, 2001, 105, 4156 CAS.
- C.-P. Tsai, S. M. Belanger, J. T. Kim, J. R. Lord and D. L. McFadden, J. Phys. Chem., 1989, 93, 1916 CrossRef CAS.
- R. T. Bise, H. Choi and D. M. Neumark, J. Chem. Phys., 1999, 111, 4923 CrossRef CAS.
- G. P. Smith, R. A. Copeland and D. R. Crosley, J. Chem. Phys., 1989, 91, 1987 CrossRef CAS.
- K. D. Hensel and J. M. Brown, J. Mol. Spectrosc., 1996, 180, 170 CrossRef CAS.
|
| This journal is © the Owner Societies 2002 |
Click here to see how this site uses Cookies. View our privacy policy here.