Temperature-induced gelation of emulsions stabilised by responsive copolymers: A rheological study
Andrew Y. C.
Koh
a,
Clive
Prestidge
b,
Igor
Ametov
b and
Brian R.
Saunders
*a
aColloid and Polymer Chemistry Research Group, Department of Chemistry, University of Adelaide, SA 5005, Australia
bIan Wark Research Institute, University of South Australia, Mawson Lakes, SA 5095, Australia
First published on 18th December 2001
Abstract
The steady-state and dynamic rheological properties of 1-bromohexadecane-in-water emulsions stabilised by responsive poly(N-isopropylacrylamide)-co-poly(ethyleneglycol methacrylate)
(poly(NIPAM-co-PEGMa)) copolymer have been investigated. The data were compared to measurements performed using dilute and concentrated poly(NIPAM-co-PEGMa) copolymer solutions. These solutions exhibit viscosities that decrease and increase, respectively, with increasing temperature. The increase in viscosity for the concentrated solution is attributed to transient network formation. The presence of dispersed oil droplets (volume fraction![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =0.30) in the emulsion causes temperature-induced gelation even though the copolymer concentration in the aqueous phase is such that transient network formation due to overlapping copolymer chains throughout the aqueous phase does not occur. Dynamic rheological data confirmed
gelation for the emulsions at elevated temperatures. Emulsion gelation is attributed to flocculation between neighbouring droplets. The adhesive droplets form a network that entraps the aqueous phase. The strength of the network increased with increasing temperature. To our knowledge these data represent the first comprehensive rheological investigation of reversible, temperature-induced gelation for an oil-in-water emulsion.
=0.30) in the emulsion causes temperature-induced gelation even though the copolymer concentration in the aqueous phase is such that transient network formation due to overlapping copolymer chains throughout the aqueous phase does not occur. Dynamic rheological data confirmed
gelation for the emulsions at elevated temperatures. Emulsion gelation is attributed to flocculation between neighbouring droplets. The adhesive droplets form a network that entraps the aqueous phase. The strength of the network increased with increasing temperature. To our knowledge these data represent the first comprehensive rheological investigation of reversible, temperature-induced gelation for an oil-in-water emulsion.
Introduction
Koh and Saunders recently reported that oil-in-water (O/W) emulsions stabilised by responsive copolymers of poly(N-isopropylacrylamide)-co-poly(ethyleneglycol methacrylate) (poly(NIPAM-co-PEGMa)) exhibited reversible, temperature-induced gelation.1 This behaviour is unusual for O/W emulsions since it is more common for these emulsions to phase separate upon heating.2 The report by Koh and Saunders used capillary viscometry. In the present study we extend the preliminary work and use steady-state and dynamic rheological measurements to investigate the temperature-induced gelation.The phenomenon of gelation for thermally responsive polymer solutions above the critical overlap concentration has received significant attention in recent years.3–6 The common structural element for the copolymers is a hydrophilic portion, which can be caused to become hydrophobic by increasing the temperature. One of the earliest reports of this phenomenon was by L’Alloret et al.7 who investigated the temperature dependent viscosity of poly(AMPS-g-PEO) copolymers in the presence of salt (AMPS and PEO are 2-acrylamido-2-methylpropoane sulfonic acid and poly(ethylene oxide), respectively). The viscosity enhancement observed with increasing temperature was attributed to hydrophobic microdomain formation. Related data have been reported in the absence of salt for poly(acrylic acid-co-AMPS)3 and also poly(acrylic acid-co-AMPS-co-PEO).4 More recently Durand and Hourdet5 have shown that the behaviour of the graft copolymer poly(acrylic acid-g-N-isopropylacrylamide) can be attributed to poly(NIPAM) microdomains connected through the highly soluble poly(acrylic acid) chains into a physical three-dimensional network.
Transient network theory has been invoked to explain the qualitative features of thermally induced gelation of concentrated responsive copolymer solutions.4,5 The theory was originally devised by Green and Tobolsky8 and has been more recently developed by Tanaka et al.9,10 Transient network theory envisages a model network made up of polymer chains with “sticky” end groups. The associative cross-links are hydrophobic clusters (junctions). An effective chain results when both ends of the chain are embedded within a junction that is connected to at least two other junctions. The steady shear viscosity is proportional to the number of elastically effective chains and the mean time the chain ends spend within a junction.
Several workers have reported studies on emulsions that are sensitive to the environmental conditions. Perrin et al.11,12 have investigated emulsions stabilised by amphiphilic copolymers based on poly(acrylic acid). The emulsion stability was found to be strongly dependent on the balance of hydrophilic and hydrophobic moieties within the copolymer as well as the ionic strength. Barnes and Prestidge13 have shown that the adsorbed layer thickness of PEO–PPO–PEO copolymers at the PDMS emulsion droplet interface is dependent on the extent of droplet cross-linking, i.e. the droplet rheology, which is temperature dependent. There has been a report of food emulsions stabilised by proteins, which undergo irreversible gelation upon heating.14 In addition, Philip et al. induced gelation of asphalt-in-water emulsions by addition of NaOH at room temperature.15 The gel phase for their emulsions exhibited strong volume contraction after formation. We are not aware of any reports of emulsions stabilised by responsive poly(NIPAM) copolymers. Furthermore, the observation of reversible, temperature-induced gelation for O/W emulsions based on these copolymers appears to be a new phenomenon.
In the present work, the synthesis of a thermally sensitive copolymer, poly(NIPAM-co-PEGMa), and its use in the preparation of thermally responsive emulsions are presented. The rheological properties of the emulsions are systematically investigated using steady-state and dynamic shear measurements as a function of temperature. The data are compared with those obtained for dilute and concentrated poly(NIPAM-co-PEGMa) solutions. The data show significant differences that reveal an important role for the dispersed droplets in emulsion gelation. More importantly, these rheological studies allow gel properties (e.g. onset of gelation and gel strength) to be determined and a gelation mechanism in emulsions and concentrated copolymer solutions at elevated temperatures to be proposed.
Experimental
Reagents
N-isopropylacrylamide (NIPAM, Acros Organics, 99%), poly(ethylene glycol methacrylate) (PEGMa, molecular weight=360, Aldrich) and azobis(isobutyronitrile)
(AIBN) were used as received. Reagent grade solvents were used without further purification and water was Millipore Milli-Q quality. 1-Bromohexadecane employed as the oil phase in the preparation of the emulsion was obtained from B.D.H.
Copolymer synthesis
Xue et al.16 have shown that the NIPAM has a strong tendency for homopolymerisation in the presence of other co-monomers. The conditions used for the preparation of poly(NIPAM-co-PEGMa) were specifically designed to promote copolymerisation through an excess of PEG macromonomer at the onset of copolymerisation.17 A representative preparation for poly(NIPAM-co-PEGMa) is as follows. PEGMa (1.73 g) and tert-butyl alcohol (37.5 ml) were added to a nitrogen-purged five-necked 250 ml reaction kettle equipped with a burette, Teflon stirrer blade, condenser, thermometer and nitrogen inlet. The stirred monomer solution was allowed to degas for 30 min before the mixture was heated to 80°C under nitrogen. NIPAM (3.27 g) and AIBN (0.03 g) in tert-butyl alcohol (12.5 ml) were added dropwise with stirring over 1 h. The solution
was refluxed for a further 4.5 h before it was allowed to cool and then added dropwise to 2.5 l of diethyl ether with constant vigorous stirring. The white solid that precipitated was filtered, washed, dissolved in milli-Q quality water (5 wt.%), extensively dialysed, before freeze-drying the sample, which gave a white powder (yield was around 50%). For comparison, poly(NIPAM) homopolymer was prepared according to the method by Winnik et al.18
Emulsion preparation
Sufficient time (1 day) was allowed for the copolymer to dissolve in water to ensure complete dissolution. 3.5 ml of the copolymer solution (3.6 wt.% copolymer) was placed in a 10 ml beaker before the addition of 1.5 ml of 1-bromohexadecane. The mixture was homogenised using a Silverson SL2T laboratory mixer for 5 min at 8000 rpm at room temperature. Dilution tests confirmed that the emulsion was of oil-in-water (O/W) type, i.e. the dispersed phase is 1-bromohexadecane.Physical measurements
The composition of both the homopolymer and copolymer was determined by IH NMR in CDCl3 using a Varian (300 MHz) spectrometer using methods reported elsewhere.19,20 The polydispersity index of the samples was obtained from GPC using Waters Styragel columns. THF was used as the eluent and the columns were calibrated with polystyrene standards. Phase transitions were monitored by the absorbance at 600 nm through a 1 cm sample cell referenced against milli-Q water, using a Cary 2200 UV–Vis. spectrophotometer. Samples (0.5 wt.%) were heated at a rate of 0.5°C min−1 in the thermally controlled cell holder. Photomicrographs of the emulsion were obtained using an optical microscope (Olympus, H30). The images were acquired using digital image-processing software and stored on a computer. Steady-state and dynamic rheological measurements were carried out using a stress-controlled
rheometer (Rheometrics SR-5000). A concentric cylinder measurement cell (inner cylinder diameter 32.0 mm and outer cylinder diameter 29.5 mm) was used for the copolymer solutions and parallel plate geometry (43 mm diameter) used for the emulsion samples. Sample temperatures during rheological analysis were controlled to ±0.1°C, through a combination of a Peltier element and an oil bath. Dynamic measurements involved the determination of the elastic (G′) and viscous moduli (G″) as a function of applied stress at an oscillation frequency of 1 Hz. G′ and G″ are linked by the phase angle (δ), i.e. tanδ=G″/G′.
Results and discussion
Copolymer preparation and characterization
A range of poly(NIPAM-co-PEGMa) copolymer compositions were screened prior to deciding upon the optimum copolymer composition for this work. A key criterion for assessing the suitability of the copolymer was its ability to stabilise O/W emulsions at room temperature whilst achieving thermally induced gelation. Poly(NIPAM) and poly(PEGMa) homopolymers did not stabilise O/W emulsions at any temperature. Poly(NIPAM-co-PEGMa) copolymers containing ca. 25, 50 and 75 mol% of PEG were synthesised and evaluated. The poly(NIPAM-co-PEGMa) copolymer containing ca. 50 mol% PEG yielded emulsions with optimum stability at room temperature whilst exhibiting thermally induced gelation behaviour.
Table 1 shows characterisation data for the poly(NIPAM-co-PEGMa) copolymer used in this work. Data for a reference poly(NIPAM) homopolymer are also shown for comparison. Obtaining good quality GPC data is challenging for poly(NIPAM) polymers. These polymers may exhibit filtration problems prior to GPC as a result of chain entanglement.21 This problem has led some workers22 to claim that GPC cannot be used to obtain molecular weight data for poly(NIPAM) polymers. As can be seen from Table 1, the poly(NIPAM-co-PEGMa) copolymer has a high polydispersity. A repeat synthesis yielded a poly(NIPAM-co-PEGMa) copolymer with the same composition and comparable Mn
(44600 g mol−1) but a lower polydispersity (2.9). Both copolymers behaved similarly
with respect to their ability to confer thermally induced gelation on oil-in-water emulsions. The polydispersity of poly(NIPAM-co-PEGMa) copolymers prepared by the synthetic method used in this work does not significantly affect emulsion behaviour. The data discussed in the remainder of the work concerns the poly(NIPAM-co-PEGMa) copolymer identified in Table 1.
Investigation of responsive copolymer solutions
Fig. 1 shows the variation of the turbidity (τ) with temperature for poly(NIPAM) and poly(NIPAM-co-PEGMa) solutions (0.5 wt.%). Values for the lower critical solution temperature (LCST) (Table 1) were taken as the point of inflection on the curves in Fig. 1, i.e., the point at which d2τ/dT2=0. The LCST for poly(NIPAM-co-PEGMa) is ca. 4.0°C higher than the value for poly(NIPAM) and is in accord with other work for related copolymers.23 It is well established that an increase in the hydrophilicity of the repeat unit by structural modification increases the LCST of poly(acrylamide)s. The fact that the LCST for poly(NIPAM-co-PEGMa) is higher than that for
poly(NIPAM) supports the view that the PEG macromonomer units are well dispersed along the copolymer chains. A block of poly(NIPAM-b-PEG) copolymer would be expected to have a significantly sharper LCST with a value closer to that for poly(NIPAM).
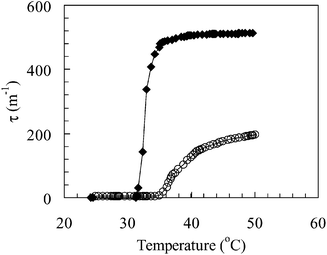 | ||
| Fig. 1 Variation of the turbidity with temperature for poly(NIPAM) (♦) and poly(NIPAM-co-MPEG) (○). | ||
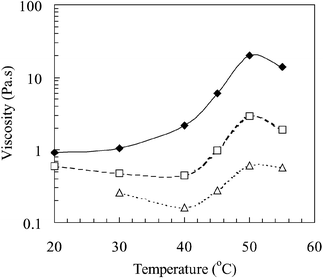
Fig. 2(a) and (b) show the variation of the viscosity (at three different shear rates) with temperature for poly(NIPAM-co-PEGMa) solutions containing, respectively, 5 and 20 wt.% copolymer. At 5 wt.% copolymer the viscosity decreases with increasing temperature; this result is expected for polymer coils below the critical overlap concentration. Temperature-induced coil collapse occurs resulting in a decrease in the disperse phase volume fraction.
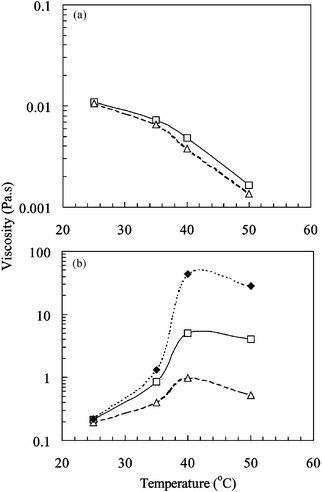 | ||
| Fig. 2 The effect of temperature on the viscosity of poly(NIPAM-co-PEGMa) copolymer solutions. The concentrations employed were 5 (a) and 20 wt.% (b). The three shear rates used were 1 (♦), 10 (□) and 100 s−1 (△). | ||
In contrast, at 20 wt.% copolymer there is evidence of temperature induced thickening between 25 and 40°C. This is attributed to temperature-induced network formation4 and is an indication that the copolymer chains overlap throughout the solution (i.e., polymer concentration greater than c*). The effect of increasing the temperature is to increase the number of elastically effective chains and also the time over which junctions containing two (or more) chains exist. There is evidence for a decrease in viscosity at temperatures greater than 40°C. This type of behaviour has been reported elsewhere4 for related copolymer solutions.
The effect of shear stress on the viscosity of 10 and 20 wt.% copolymer solutions is shown in Fig. 3(a) and (b), respectively. (5 wt.% copolymer solutions exhibited viscosities less than 0.0050 Pa s with poor signal-to-noise ratios and are not presented here). The data indicate a temperature-induced association effect is operative above the LCST. An increase in the temperature increases the number and longevity for elastically effective chains. The effect becomes more pronounced at increasing polymer concentration because of a greater proportion of elastically effective chains.
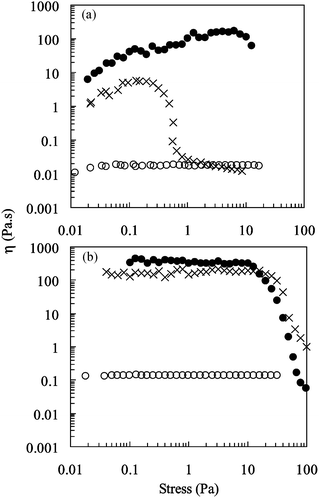 | ||
| Fig. 3 Viscosity as a function of shear stress for poly(NIPAM-co-PEGMa) copolymer solutions. The copolymer concentrations employed were 10 (a) and 20 wt.%
(b). The temperatures investigated were 25 (○), 40 (×) and 50°C (•). | ||
Fig. 4 and 5 show dynamic rheological data for a 20 wt.% copolymer solution measured at different temperatures. Both the elastic modulus (G′) values and the range of linear viscoelasticity (i.e. stress range where G′ is independent of stress) are considerably greater at temperatures higher than the LCST, supporting the view that an increase in temperature produces a viscoelastic network. Fig. 5 indicates that the phase angle (δ) approaches 45°
(i.e.G′=G″) at elevated temperatures, which again supports gelation. (At 25°C the solution behaves as a viscous liquid (G′≪G″)). It is noted that the term gelation is used here to describe the reversible formation of non-covalent, associative,
bonds. The primary modes of attraction for the groups involved are van der Waals and hydrophobic forces. This differs considerably from the covalently bonded networks studied by Mortimer et al.24 The polymer concentration required for a significant temperature-induced gelation effect is ∼10 wt.% for the copolymer solutions investigated in this work.
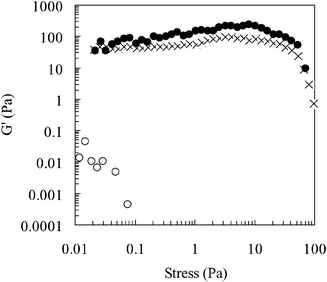 | ||
| Fig. 4 The storage modulus (G′) as a function of shear stress for a 20 wt.% poly(NIPAM-co-PEGMa) copolymer solution. The temperatures investigated were 25 (○), 40 (×) and 50°C (•). | ||
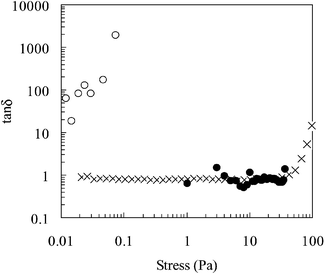 | ||
| Fig. 5 Tanδ as a function of shear stress for a 20 wt.% poly(NIPAM-co-PEGMa) copolymer solution. The temperatures investigated were 25 (○), 40 (×) and 50°C (•). | ||
Investigation of responsive emulsions
Poly(NIPAM-co-PEGMa) copolymer was found to be an effective O/W emulsifier for 1-bromohexadecane-in-water emulsions. Fig. 6 shows representative optical micrographs for an emulsion prepared using an oil phase volume fraction (ϕ0) of 0.30. These were obtained prior to (Fig. 6(a)) and after (Fig. 6(b)) one temperature-induced gelation cycle, i.e., heating from room temperature to 50°C and holding at the temperature for ca. 30 min and then cooling to room temperature. It is evident that the droplets are reasonably polydisperse and weakly flocculated. A droplet size distribution was obtained from the data shown in Fig. 6(a) and fitted to log–normal distribution25 profile. The values for the peak diameter
and broadness parameter were 7.2 µm and 0.42, respectively. These data for the emulsion together with its weakly flocculated nature (prior to gelation) are typical of emulsions stabilised by macrosurfactants.11 The values for the peak diameter and broadness parameter were 9.6 µm and 0.39, respectively, for the emulsion shown in Fig. 6(b). The effect of temperature on the storage stability of the emulsions will be the subject of a future publication. It is important to note that the flocs observed at room temperature in Fig. 6(a) could be broken up with gentle agitation. Electrophoresis measurements on the droplets at room temperature revealed a zeta potential values less than 5 mV. This low value suggests that the emulsion stability at room temperature is due to steric rather than electrostatic interaction. The extent of steric stabilisation will diminish
as the solution temperature approaches the LCST (and θ-temperature) of the copolymer.26 Kapsabelis and Prestidge27 revealed that the adsorbed layer thickness for cellulose-based polymers was highly temperature sensitive, but that adsorbed layer contraction was dependent on a balance of the segment–solution, segment–segment and segment–surface interaction parameters.
 | ||
| Fig. 6 Optical micrograph of the emulsion at room temperature: before (a) and after (b) gelation. | ||
Fig. 7 shows the variation of emulsion viscosity with temperature, as determined at shear rates of 1, 10 and 100 s−1. The viscosity data at the higher shear rate (100 s−1) are in agreement with data measured for related emulsions using capillary viscometry.1 The variation of the emulsion viscosity with temperature in Fig. 7 for the lowest shear rate (1 s−1) shows similar behaviour to the equivalent data shown in Fig. 2(b) for the concentrated copolymer solution. Thus, pronounced thickening of the emulsion occurs at low shear rates once the temperature exceeds the LCST. Sample tubes containing the emulsion can be inverted at 50°C with the gelled emulsion remaining in its original position.1 Overall, the dispersed droplets appear to have the same
qualitative effect on the rheological properties as a substantial increase in the copolymer concentration, i.e., addition of the oil phase results in a thermally-induced gelation which would have otherwise been absent at that temperature.
 | ||
| Fig. 7 The effect of temperature on the viscosity of a 1-bromohexadecane-in-water emulsions prepared using poly(NIPAM-co-PEGMa). The three shear rates used were 1 (♦), 10 (□) and 100 s−1 (△). | ||
Fig. 8 shows the effect of shear stress and temperature on the viscosity of the emulsion. The emulsion exhibits weak shear-thinning behaviour at 25°C. This may be ascribed to droplet deformation, which is a well-known phenomenon for emulsions under shear28 and the disruption of weak flocs. Upon increasing the temperature a significant increase is observed in the low stress viscosity and the stress required for the onset of thinning (i.e. the apparent yield stress). This rheological behaviour is indicative of the formation of a transient network upon heating. The number of effective chains and the amount of time that junctions remain in hydrophobic clusters will increase with temperature, i.e. a temperature increase results in a stronger network. The network is considered to be based upon flocculated emulsion droplets, where the droplets are held together by attractive hydrophobic interactions
between clusters of copolymer segments at the interface, as well as van der Waals attraction between the droplets themselves. Increasing the temperature results in a stronger hydrophobic attraction and therefore a greater shear stress is required to disrupt the junctions.
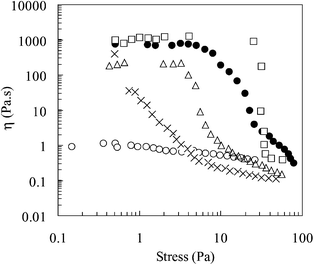 | ||
| Fig. 8 The effect of shear stress on the viscosity of 1-bromohexadecane-in-water emulsions prepared using poly(NIPAM-co-PEGMa). The measurement temperatures were 20 (○), 40 (×), 45 (△), 50 (•) and 60°C (□). | ||
The steady state rheological data for the emulsion (e.g.Fig. 8) may also be considered in terms of viscosity (η) vs. shear rate (dγ/dt) and, in the shear-dependent regime, can be described in terms of a power-law model:29
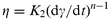 | (1) |
ηvs. ln(dγ/dt) plots (i.e. where the viscosity was significantly less than the zero shear viscosity) and are plotted against temperature in Fig. 9
(R2 values for the fits were greater than 0.994). The values for n are less
than unity and are consistent with shear thinning. The K2 values relate directly to the structure of the emulsion droplet network and exhibit a pronounced increase above the LCST for the copolymer. A concomitant decrease in n is observed; this reflects a greater extent of shear thinning for the more highly structured emulsions.
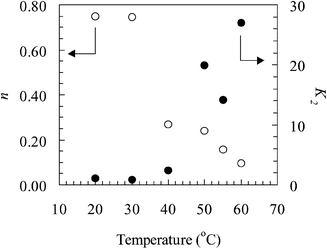 | ||
| Fig. 9 The effect of temperature on the values of n (○) and K2 (●) obtained from fitting the power-law model (eqn. (1)) to the viscosity vs. shear rate data for the emulsion. | ||
The dynamic rheological properties of the emulsion were also determined as a function of temperature: Fig. 10 and 11 show, respectively, the variation of G′ and tanδ with shear stress. The observed increase in G′ with temperature is indicative of an increased elasticity of the emulsion29 and is consistent with network formation. The values for tanδ decrease with increasing temperature, which further supports the transformation from a viscous fluid to an elastic, gel-like structure. It should also be noted that tanδ values for the emulsion are less than that for the equivalent copolymer solution, again reflecting differences in microstructure and gel properties.
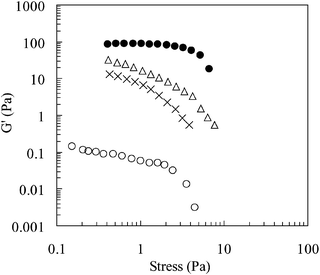 | ||
| Fig. 10 The storage modulus (G′) as a function of shear stress for a 1-bromohexadecane-in-water emulsions prepared using poly(NIPAM-co-PEGMa). The temperatures were 20 (○), 40 (×), 45 (△), 50°C (●). | ||
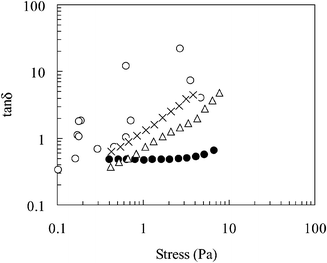 | ||
| Fig. 11 Variation of tanδ as a function of shear stress for a 1-bromohexadecane-in-water emulsions prepared using poly(NIPAM-co-PEGMa). The temperatures were 20 (○), 40 (×), 45 (△), 50°C (●). Note that the variability of the data measured at 20°C is due to the low values of G′ and G″. | ||
Proposed mechanism for temperature-induced emulsion gelation
The mechanism for the observed temperature-induced gelation of the emulsion is of considerable interest. There are several key experimental observations that must be accounted for: (a) The prevention of phase separation as a consequence of gelation. (b) The relatively high degree of reversibility for the gelation transition, and (c) The observation of temperature-induced gelation for emulsions containing a continuous phase copolymer concentration less than that required for gelation of the parent copolymer solution.The consequence of increasing the temperature to above the LCST for the emulsion is considered to be a change from repulsive to attractive inter-coil interactions.26 This will necessarily cause droplet–droplet interactions to become attractive, resulting in flocculation. The adsorbed copolymer chains will also collapse leading to a more rigid interfacial layer to oppose coalescence. This would account for the prevention of phase separation upon gelation and increase in elastic structure. It follows that a finite contact angle should emerge between droplet pairs as the temperature increases. This aspect will be investigated in future work.
Strongly flocculated emulsions invariably consist of aggregates containing considerable entrapped solvent.30 The flocculated droplets will form a three-dimensional network provided that a critical ϕ0 is exceeded such that chains of droplets can achieve a sufficient degree of connectivity throughout the emulsion volume. As the temperature decreases, the collapsed polymer chains begin to expand (as now water becomes a good solvent). This causes the network to break-up and the flocculated droplets that once formed the three-dimensional network, to redisperse.
It is appropriate to comment on the complex form of the attractive interactions that are believed to maintain the gelled state for the emulsions. The three-dimensional droplet network structure would consist of droplet chains held together by overlapping copolymer chains adsorbed at the emulsion interface. (A schematic representation is given in Fig. 12). These interfacial copolymer chains form their own transient network, which acts as the “glue” for the droplet network. Thus, only copolymer adsorbed at the droplet surface need be involved in formation of the connected droplet network and gelation. An attractive feature of this model is that a gelled network can result in the presence of a lower copolymer concentration than the critical overlap concentration. An entangled mesh of copolymer chains existing throughout the aqueous phase is not required for emulsion gelation.
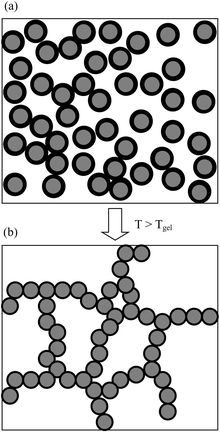 | ||
| Fig. 12 Diagram depicting the proposed mechanism for thermally-induced gelation for an O/W emulsion. The oil droplets (grey) contain an adsorbed layer of copolymer (black) in each case. At room temperature (a) the layer is expanded and at elevated temperature (b, above the LCST) it is collapsed and the emulsion gels. | ||
Conclusions
This work has provided a detailed rheological investigation into a new type of emulsion behaviour: temperature-induced emulsion gelation. It has been clearly shown that the presence of a dispersed oil phase (ϕ0=0.30) within a dilute copolymer solution results in temperature-dependent rheological properties that are significantly different to that of the parent copolymer solution. The emulsion exhibits temperature-induced gelation that is mostly reversible, whereas, the viscosity of the dilute copolymer solution decreases with increasing temperature. The gelled emulsions are sensitive to shear and the gel-to-fluid transition results at modest shear stresses (<100 N m−2). The mechanism proposed to explain the temperature-induced gelation envisages bridging flocculation as a consequence of an increase in the polymer–solvent interaction parameter (to greater than 0.5) for the stabilising
copolymer chains with increasing temperature. Above the LCST, the copolymer–copolymer interactions become attractive resulting in transient network formation between neighbouring droplets and creation of networks of flocculated droplets.
It is important to note that reversible, temperature-induced gelation is a general effect for emulsions stabilised by poly(NIPAM-co-PEGMa). We have observed equivalent behaviour for a range of oil phases including toluene, poly(dimethylsiloxane) and perfluorodecalin. This reversible gelation process may be of great technological importance in the emulsion field because it allows the transformation of a fluid emulsion into a gel in a short time using temperature alone as the trigger.
Acknowledgements
The authors would like to thank the University of Adelaide for providing an international scholarship to A.K. We also thank Dulux Australia for providing the GPC data. A University of Adelaide Grant is acknowledged. C.P. and I.A. acknowledge funding from the Australian Research Council’s Special Research Centre for Particle and Material Interfaces.References
- A. Koh and B. R. Saunders, Chem. Commun., 2000, 2461 RSC.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 2539 CrossRef CAS.
- A. Durand and D. Hourdet, Macromol. Chem. Phys., 2000, 201, 858 CrossRef CAS.
- F. L'Alloret, P. Maroy, D. Hourdet and R. Audebert, Rev. Inst. Fr. Pet., 1997, 52, 117 Search PubMed.
- A. Durand and D. Hourdet, Polymer, 1999, 40, 4941 CrossRef CAS.
- A. M. Mathur, B. Drescher, A. B. Sevanton and J. Klier, Nature, 1998, 392, 367 CrossRef CAS.
- F. L'Alloret, D. Hourdet and R. Audebert, Colloid Polym. Sci., 1995, 273, 1163 CrossRef CAS.
- M. S. Green and A. V. Tobolsky, J. Chem. Phys., 1946, 14, 80 CrossRef CAS.
- F. Tanaka and S. F. Edwards, Macromolecules, 1992, 25, 1516 CrossRef CAS.
- F. Tanaka and M. Ishida, Macromolecules, 1996, 29, 7571 CrossRef CAS.
- P. Perrin and F. Lafuma, J. Colloid Interface Sci., 1998, 197, 317 CrossRef CAS.
- P. Perrin, N. Monfreux and F. Lafuma, Colloid Polym. Sci., 1999, 277, 89 Search PubMed.
- T. J. Barnes and C. A. Prestidge, Langmuir, 2000, 16, 4116 CrossRef CAS.
- E. Dickinson and S.-T. Hong, Colloids Surf. A, 1997, 127, 1 CrossRef CAS.
- J. Philip, L. Bonakdar, P. Poulin, J. Bibette and F. Leal-Calderon, Phys. Rev. Lett., 2000, 84, 2018 CrossRef CAS.
- W. Xue, S. Champ and M. B. Huglin, Polymer, 2000, 41, 7575 CrossRef CAS.
- A. E. Cardenas-Valera, A. I. Bailey and A. Doroszkowski, Colloids Surf. A, 1995, 96, 53 CrossRef CAS.
- F. M. Winnik, Macromolecules, 1990, 23, 233 CrossRef CAS.
- X. Qiu and C. Wu, Macromolecules, 1997, 30, 7921 CrossRef CAS.
- F. Zeng, Z. Tong and H. Feng, Polymer, 1997, 38, 5539 CrossRef CAS.
- F. Ganachaud, M. J. Monteiro, R. G. Gilbert, M.-A. Dourges, S. H. Thang and E. Rizzardo, Macromolecules, 2000, 33, 6738 CrossRef CAS.
- X. Y. Wu, R. H. Pelton, K. C. Tam, D. R. Woods and A. E. Hamielec, J. Polym. Sci., Part A, 1993, 31, 957 Search PubMed.
- J. Virtanen and H. Tenhu, Macromolecules, 2000, 33, 5970 CrossRef CAS.
- S. Mortimer, A. J. Ryan and J. L. Stanford, Macromolecules, 2001, 34, 2973 CrossRef CAS.
- Y. De Smet, D. Danino, L. Deriemaeker, Y. Talmon and R. Finsy, Langmuir, 2000, 16, 961 CrossRef CAS.
- D. H. Napper, Polymeric Stabilization of Colloidal Dispersions, Academic Press, New York, 1983. Search PubMed.
- S. Kapsabelis and C. A. Prestidge, J. Colloid Interface Sci., 2000, 228, 297 CrossRef CAS.
- P. Manoj, A. D. Watson, D. J. Hibberd, A. J. Filler-Travis and M. M. Robins, J. Colloid Interface Sci., 1998, 207, 294 CrossRef CAS.
- H. A. Barnes, J. F. Hutton and K. Walters, An Introduction to Rheology, Elsevier, Amsterdam, 1989..
- H. M. Princen, M. P. Aronson and J. C. Moser, J. Colloid Interface Sci., 1980, 75, 246 CrossRef CAS.
| This journal is © the Owner Societies 2002 |