Coupling of solvent and solute dynamics—molecular dynamics simulations of aqueous urea solutions with different intramolecular potentials
Received 2nd July 2001, Accepted 11th October 2001
First published on 10th December 2001
Abstract
The vibrational characteristics and the hydration of urea are studied by classical molecular dynamics simulations, using three different urea models that are dissolved in 1616 SPC/E waters. The urea models only differ in the frequency of nitrogen inversion, ranging from 645 to 170 cm−1 in the gas phase. Intermolecular interactions are described by additive potentials using the same parameters for all models. The effects of urea on local water properties are discussed in terms of radial profiles of excess coordination numbers, the extent of the hydrogen-bond network, and the water–water interaction energy. It is found that water properties beyond the first hydrate shell of an urea model with slow inverting pyramidal amino groups are similar to those beyond the second hydrate shell of a water molecule, whereas a planar quickly vibrating urea model induces perturbations similar to hydrophobic hydration. The results are discussed in the context of the
usefulness of molecular dynamics simulations for the understanding of the denaturing effect of urea on water.
1 Introduction
The structural and dynamical features of urea–water systems gained much attention during the last thirty years because of their importance for the understanding of liquid water itself. Observed effects like denaturing of water soluble polymers or increased solubility of hydrocarbons in water when urea is added led to the popular view of urea being a “structure breaker”. Frank and Franks1 explained these effects by a change of the equilibrium between disordered dense and ice-like water in the presence of urea. Stokes,2 Schellman3 and Kreschek and Scheraga4 favoured the formation of urea oligomers at higher concentrations as a possible reason for the special behaviour of urea–water mixtures.Various simulations by means of molecular dynamics (MD) and Monte Carlo (MC) techniques were reported,5–17 which analysed the thermodynamical, structural and dynamical properties of these systems in order to aid the interpretation of innumerable experiments and to prove one or other model. Because different potentials for intermolecular interactions and different measures of water structure and dynamics were used, controversial conclusions regarding the effect of urea on water were published. The reported analyses of structural properties of urea–water mixtures point to a near ideal fit of urea into liquid water,6,7,12,14,17 thus no significant disturbance of water structure and no tendency to form aggregates at lower concentrations was observed. This interpretation disagrees with studies of dynamical properties.
Tovchigrechko et al.15 reported an increased mobility of water behind the first hydration shell of urea. They combined their computation of transport coefficients with analyses of the network structure of water and concluded that urea leads to network defects of water by increasing the fraction of mobile low-energetic five-coordinated water molecules. In contrast to that, Idrissi et al.16 reported intermolecular vibrational spectra of water in urea–water mixtures from MD simulations. They interpreted their data in terms of stiffening of translational water dynamics when urea is added.
All previous studies used carefully developed intermolecular potentials, but paid no attention to the intramolecular dynamics of urea itself. Since most MD and MC simulations used parameters from standard force fields like GROMOS or AMBER for intramolecular degrees of freedom, they described urea to behave like common amides. These force fields define amides to contain a planar CONH2 moiety by applying appropriate out-of-plane and torsion terms. By using these parameters for urea, it shows C2v symmetry. Indeed, several neutron diffraction studies18–21 of crystalline urea confirmed that it has a planar structure in the solid state. On the other hand, from gas phase spectra22,23 it was proposed that the potential energy minimum structure of isolated urea is a C2 structure with pyramidal amino groups. This result is also supported by a number of
high-level quantum mechanical studies.23–27 It was also demonstrated that urea-water clusters can be found showing urea nitrogen as hydrogen bond acceptor.28,29 These findings indicate that previous studies on urea–water mixtures by means of molecular mechanics suffer from a correct description of the intramolecular potential of the solute, which is coupled with the neglect of important specific solute–solvent interactions.
However, the importance of the pyramidality of the amino groups of urea and their ability to form hydrogen bonds for the structure and dynamics of water in its aqueous solution depends on the timescale of their inversion. Raman spectra of pure liquid water show two bands at about 60 and 170 cm−1, which have to be interpreted as intermolecular vibrations of water molecules in a hydrogen-bonded network.30 Hydrogen bond lifetimes in pure water are estimated from MD simulations to amount to about 0.5–1 ps.31 If nitrogen inversion of urea is on the same time scale or slower, it should be very well possible that the structure and dynamics of its hydrate shell is different from that formed around quickly vibrating planar urea. So it is fair to conclude that a wrong description of the intramolecular vibrational behaviour of urea causes wrong conclusions regarding its effect on water properties. The only experimental
information about the timescale of inversion of the amino groups of urea is provided by the low-temperature matrix isolation infrared spectrum of King.32 He observed a band at 227 cm−1, which he interpreted as the NH2 wagging mode or “inversion”. A number of ab initio or density functional theory (DFT) calculations23,33–35 on the vibrational characteristics of urea in the gas phase indicate the lowest normal modes above 350 cm−1. Godfrey et al.23 discussed this discrepancy by means of the complicated potential energy surface along the reaction coordinate of N-inversion. It turns out that N-inversion does not correspond to a fundamental vibration. It is rather a conformational change, which has to be characterized by an energy of activation and the corresponding rate constant. According to their data,
the difference of potential energies of the transition state of inversion and a C2 structure of gaseous urea amounts to 1.6 kcal mol−1. After applying the Eyring equation at room temperature, N-inversions should occur with a rate constant of about 5 ps−1, or a frequency below 200 cm−1. Taking the uncertainty of these estimations into account, the presence of an inversion vibration in the low-temperature IR spectrum is probably ruled out, but additional evidence for the importance of this event regarding coupling of solvent and solute dynamics is provided. Unfortunately, there exist no informations about the conformational behaviour of urea in water.
In order to understand whether and how the flexibility of the amino groups of urea affects properties of its surrounding solvent, we describe MD simulations of diluted urea solutions using three different urea models. The models define similar parameters for intermolecular interactions, which were derived from geometries and interaction energies of urea–water complexes calculated with a density functional method. The intramolecular potentials of these models are varied in order to scale the frequency of N-inversion in the gas phase from 170 to about 640 cm−1. The effects of this scaling on water properties are analysed by means of comparison of local water densities, energetics and dynamics in different parts of the urea solution with those in pure water.
2 Methods and systems
2.1 Potentials
We decided to use an all-atom effective potential with harmonic or periodic expressions for intramolecular degrees of freedom and a Coulomb and Lennard-Jones pair-potential for non-bonding interactions:| | 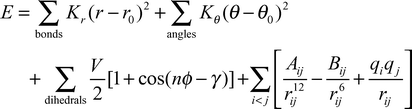 | (1) |
We used the rigid SPC/E water model36 along with three different urea models. All urea models define the same Lennard-Jones parameters and fixed atomic charges (see Table 1). The charges were produced by fitting them to the electrostatic potential of urea calculated with the HF/6-31G* method according to the Merz–Singh–Kollman scheme.37 This approach yields a permanent dipole moment of the urea models of 4.4 to 4.7 D† in aqueous solution, depending on the
intramolecular potentials. It has to be compared with 4.56 D measured in dioxane,38 and with values ranging from 2.8 D6 in earlier MD simulations to 4.4 D16 as used in a recent study. Lennard-Jones parameters were taken from the AMBER 4.039 force field and adjusted to yield satisfactory potential energies of urea–water complexes, which were compared with those obtained with the Becke3LYP/6-31G* density functional method40
(see Fig. 1).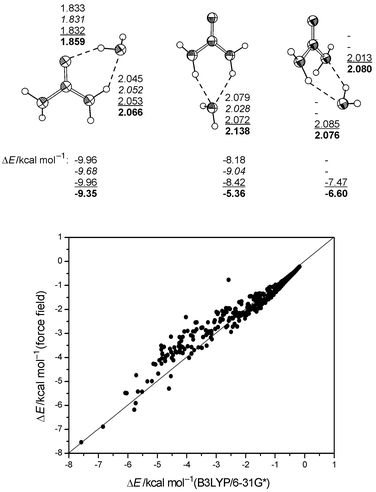 |
| | Fig. 1 Top: H-bond distances (Å) and association energies of stable urea–water complexes (U1, U2, U3, B3LYP/6-31G* corrected for BSSE). Bottom: Comparison between uncorrected interaction energies of 450 urea–water complexes (urea constrained to geometry from B3LYP/6-31G*, OW constrained to grid points of a 3D grid around urea up to 6 Å from urea, water orientation optimized). | |
Table 1 Non-bonded parametersa
| Atom | R*/Å | ε/kcal mol−1 | q/e |
|---|
| Potential see eqn. (1). Note that Aij
=
εij(Rij*)12, Bij
= 2εij(Rij*)6, εij
=
(εiεj)1/2, Rij*
=
Ri*
+
Rj* for interaction of atom i and atom j. |
|---|
| C | 1.9080 | 0.0860 | 1.0950 |
| O | 1.7683 | 0.2100 | −0.6654 |
| N | 1.8240 | 0.1700 | −1.0810 |
| Hcis | 0.6000 | 0.0150 | 0.4273 |
| Htrans | 0.6000 | 0.0150 | 0.4389 |
| OW | 1.7766 | 0.1554 | −0.8476 |
| HW | 0.0000 | 0.0000 | 0.4238 |
Intramolecular parameters of urea were taken initially from the AMBER 4.0 force field. They were then parameterized on the frequencies of the lower normal modes, which were calculated in the harmonic approximation. In addition, the scaling factor of 1–4 electrostatic interactions was varied in order to obtain the desired vibrational behaviour regarding inversion of the amino groups. The used parameters of three urea models are summarized in Tables 2 and 3. Unscaled frequencies of fundamental vibrations are shown in Table 4. A detailed description of the resulting vibrational characteristics of the urea models will be given in section 3.
Table 2 Angle parametersa
| Angle | θ0/° | K/kcal mol−1 rad−2 |
|---|
| U1 | U2 | U3 | U1 | U2 | U3 |
|---|
| Potential see eqn. (1). |
|---|
| O–C–N | 122.9 | 121.4 | 122.9 | 80.0 | 100.0 | 80.0 |
| N–C–N | 114.2 | 117.2 | 114.2 | 80.0 | 80.0 | 80.0 |
| C–N–H | 120.0 | 120.0 | 114.7 | 30.0 | 30.0 | 40.0 |
| H–N–H | 120.0 | 120.0 | 113.6 | 35.0 | 35.0 | 30.0 |
Table 3 Proper and improper torsional parametersa
| Torsion | (V/2)/kcal mol−1 | γ/° | n |
|---|
| U1 | U2 | U3 |
|---|
| Potential see eqn. (1), 1–4 vdW-interactions scaled with 0.5, 1–4-electrostatics scaled with 0.5 (U1, U3), 0.833 (U2). |
|---|
| x–C–N–x | 2.5 | 2.5 | 2.0 | 180.0 | 2 |
| O–C–N–H | 2.5 | 2.5 | 2.5 | 180.0 | 2 |
| | 2.0 | 2.0 | 2.0 | 0.0 | 1 |
| |
| x–x–C–O | 10.5 | 15.0 | 15.0 | 180.0 | 2 |
| x–x–N–H | 1.0 | — | — | 180.0 | 2 |
2.2 Systems and simulation procedure
The systems described here contain one urea molecule and 1616 SPC/E water molecules in a rectangular box, corresponding to an ideal solution with a concentration of 0.035 M. Previous simulations used up to 350 water molecules. We decided to extend the systems on the basis of observations on the long-range behaviour of the urea–water pair potential, which made a cutoff of non-bonding interactions larger than 10 Å necessary in order to reduce artefacts from neglecting long-range electrostatics, and to obtain a substantial amount of bulk water in the simulation box.The MD simulations started with pre-minimized structures. The systems were equilibrated for 100 ps at 300 K and 1 atm, followed by 1 ns trajectories for data collection. Newton's equations of motion were integrated using the half-step “leap-frog” scheme37 of the Verlet algorithm with an integration time of 1 fs. The time evolution was recorded every 10 fs. Constant temperature dynamics was performed by scaling atomic velocities according to Berendsen et al.41 using a time constant of 0.1 ps. The coupling to a pressure bath described by Berendsen et al.41 used a time constant of 1 ps. Interaction energies were cut with a hard cutoff at 12 Å. The non-bonded pair list was recalculated every 10 fs. Gas phase simulations of isolated urea were performed at 300 K for 300 ps, taking snapshots every 1 fs. The molecular translational and rotational movements were annihilated
every 25 fs.
Ab initio and DFT calculations as well as characterizations of stationary points of the force field models were carried out with the Gaussian98 A5 program,42 which we modified to contain the necessary force field parameters and routines. MD simulations were performed with the AMBER 5.0.1 package.43 For visualization we used SYBYL 6.6.44 All results were produced on SGI Octane and SGI Origin 2000 systems.
2.3 Methods of evaluation
2.3.1 Statistics. Reported quantities A are time averages 〈A〉 over 1 ns trajectories, assuming a gaussian distribution of A. Errors σ
(〈A〉) are estimated by scaling the standard deviation σ
(A) by a factor derived from the statistical inefficiency45,46 of the data. The error of derived quantities like hydration enthalpies etc. is obtained from error propagation. 2.3.2 Local water properties. Properties of water like density, self-energy or hydrogen bond characteristics were calculated in different regions around the solute. Properties of bulk water were extracted from these data as averages from 12 to 16 Å from the centre of mass of urea, rather than using an independent simulation of pure water or averaging over the whole simulation boxes.Pair distribution functions gXS(PDF, X = solute, S = solvent) in volume elements dV were defined according to
| | 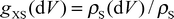 | (2) |
where
ρS(d
V) is the number density of water atoms or molecules in d
V defined in a solute-fixed coordinate system, and
ρS is the number density of water in the simulation box. By defining d
V as a spherical shell at increasing distance
r from X, the usual radial distribution function
gXS(
r)
(RDF) is obtained. By defining cubic volume elements in a 3D-grid and plotting resulting iso-probability surfaces of solvent atom positions, the peaks of the RDF's can be resolved in space. The cartesian coordinate system used defines the N–N direction in urea as the
x-axis, and the perpendicular of urea oxygen on
this axis as the
z-axis. Urea is nearly fixed in this coordinate system and shows time-averaged
C2v symmetry, only the amino hydrogens vibrate around the
xz plane.
The perturbation of water density is measured by the excess running coordination number ΔNXS(r),47 which is defined as the difference between the number of water molecules found around the solute in the solution and the number of water molecules found around a water molecule in pure water:
| | 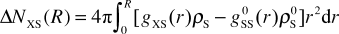 | (3) |
The superscript 0 indicates pure (bulk) water. Positive differential contributions to the integral indicate an increased local density of water compared with pure water. The same approach is used to describe local perturbations of water–water interactions Δ
ES(
r):
| | 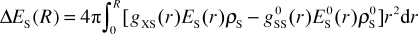 | (4) |
Here
ES(
r) is the potential energy of a solvent molecule located at distance
r from the central molecule (urea
in the case of
ES, water in the case of
E0S) in the field of all remaining solvent molecules, related to infinite separation of the considered solvent molecule and its surrounding solvent.
E0S(
r→∞) is equal to the potential energy per solvent molecule in a pure solvent system, which is similar to the molar self-solvation energy of the solvent or its negative molar vaporization energy. Expression (4) converges at large
R to the solvent reorganization energy Δ
ES, which can also be obtained as the difference between the potential energy of the solvent in the simulation box excluding solute–solvent interactions and the total potential energy of a pure solvent system containing the same number of solvent molecules.
The hydration enthalpy of a solute in water can be obtained from simulations using additive potentials by
| | 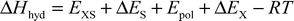 | (5) |
The major contribution to this expression is the solute–solvent interaction energy
EXS, which is obtained by summation of non-bonded potential terms of all solute–solvent pairs of one snapshot, and averaging over the trajectory. The last term of
eqn. (5) describes the work necessary for transferring one mole of solute molecules from an ideal gas state into the solvated state, whereby changes of the volume of the solution are neglected. The polarization correction
Epol represents an estimation of the energy needed to polarize the gas phase charge density of the solute to yield the dipole moment of the effective pair model. It can be computed by
36| | 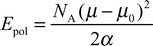 | (6) |
where
NA is Avogadro's number,
μ is the permanent dipole moment of the solute used in the force field,
μ0 is its dipole moment in the gas phase, and
α is its isotropic static polarisability. By using
μ0
= 3.5 D from MP2/6-311+G*
ab initio calculations,
α
= 6.5 × 10
−40 C
2 m
2 J
−1 from high level DFT calculations,
25 and
μ
= 4.5 D from our force field, a correction of 1.23 kcal mol
−1 is obtained. The term Δ
EX of
eqn. (5) is the change of the internal energy of the solute on solvation. We obtain it as difference of averages from MD simulations of the solution and the isolated solute.
2.3.3 Vibrational analyses. Vibrational spectra of urea were extracted from the simulations as Fourier transforms ![[D with combining tilde]](https://www.rsc.org/images/entities/i_char_0044_0303.gif) (ω) of the molecular dipole autocorrelation functions D(t)
(DACF):
(ω) of the molecular dipole autocorrelation functions D(t)
(DACF):| | 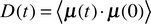 | (7) |
where μ(t) is the permanent dipole moment vector of urea at time t. We used a correlation time of 10 ps. The infrared intensity I(ω) at frequency ω is then obtained by48| | 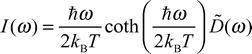 | (8) |
where kB is Boltzmann's constant and T is the absolute temperature. The assignment of bands to nuclear motions was done by analysing autocorrelation functions of the cartesian components of the dipole vector in a solute-fixed coordinate system (given
above). In addition, the vibrational behaviour of urea regarding N-inversion was characterized by trajectories, which we defined as the distance of nitrogen from the oriented plane given by its neighbouring atoms. An inversion event is recognized by a change of the sign of the trajectory. Counting of events yields an inversion frequency. Fourier transforms of the trajectories yield the same frequency, if inversions occur strictly periodically in time. Otherwise they show no or only weak and broad bands. By using counted frequencies from the trajectories, we are able to describe inversions that cannot be recognized from the IR spectra, as well as to obtain mean amplitudes of pyramidalization and lifetimes of pyramidal amino groups as additional measures for the vibrational characteristics of the solute. 3 Results and discussion
3.1 Vibrational dynamics of urea
The used parameter sets yield variations of the torsional and wagging normal modes of urea by retaining frequencies of the remaining fundamental vibrations (see Table 4). It is clear that the simple potential we used is not suitable to describe all fundamentals with good accuracy, especially the CN stretching modes need more attention. On the other hand, quick vibrations like these should have a minor effect on the intermolecular dynamics of the solution, compared with vibrations in the low-frequency region. The planar model U1 yields the δ(CN) and δ(CO) vibration as the lowest normal modes, followed by out-of-plane motions of the NH2 groups from 550 to 670 cm−1. All of them are faster than the corresponding vibrations obtained by ab initio calculations. When the improper NH torsion is omitted and N–H and O–H 1–4 electrostatics are taken
into account to a greater extent (U2), very slow NH2 out-of-plane modes are obtained, leaving the remaining spectrum mostly unchanged. By introducing XNH angles lower than 120°
(U3) all modes involving XNH angle deformations are shifted to lower frequencies. Calculated IR spectra are shown in Fig. 2. We identify the stretching vibrations involving hydrogens above 3300 cm−1, the ν(CO), δ(NH2) and the asymmetric ν(CN) between 1700 and 1500 cm−1, and the NH2 rocking and the symmetric ν(CN) from 800 to 900 cm−1 in the spectra of all models. Differences between the three urea models occur below 800 cm−1. The planar model U1 yields a sharp and intense band at 754 cm−1, which we assign to the ω(CO) mode. A second
very small band corresponding to a vibration perpendicular to the molecular plane is detected at 590 cm−1, which splits into two intensive bands in the Fourier transform of the velocity autocorrelation function (power spectrum, not shown) and which describes the inversion of amino groups. The lowest modes at 508 and 467 cm−1 are the δ(CO) and δ(CN) vibrations, as already indicated by the calculated normal vibrations (see Table 4). U2 and U3 show two weak and broad bands at about 380 and 620 cm−1. The latter describes the ω(CO) vibration already found in U1 in the same region. The former we assign to out-of-plane vibrations of the amino groups. Trajectories of N-inversion obtained for one nitrogen atom of each urea model in the gas phase are shown in Fig. 3. The resulting vibrational
characteristics are summarized in Table 5. Model U1 is vibrating very fast around a planar structure, showing only small amplitudes. The counted frequency is higher than obtained from the spectrum shown in Fig. 2, because not every counted inversion event reaches the same amplitude. The counted frequency of inversion is reduced drastically for models U2 and U3. U2 shows a slow large-amplitude periodic inversion, which is superimposed with a faster vibration of about 400 cm−1 along the reaction coordinate. This frequency is in agreement with that obtained from Fig. 2. U3 rests in pyramidal states for about 100 fs, until inversion occurs. This behaviour corresponds to a counted inversion frequency of about 170 cm−1. This model also shows a faster superimposed vibration in pyramidal states. The latter should be responsible for the band observed in the vibrational
spectrum as already discussed for U2, whereas the inversion motion is aperiodic and too slow to become detected by Fourier analysis of the DACF or the trajectory itself. In addition to these characteristics, the trajectories reveal that inversions of both amino groups of an urea molecule are strictly correlated in U2 and U3. They occur almost simultaneously and interconvert the two stereoisomers with averaged C2 symmetry into each other. In the used empirical potential, electrostatic interactions between hydrogens and nitrogen should be responsible for this behaviour.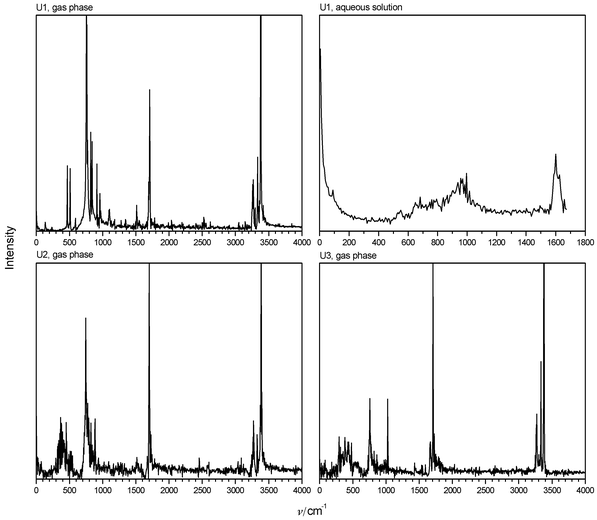 |
| | Fig. 2 IR spectra of urea calculated from autocorrelation functions of the urea dipole. | |
 |
| | Fig. 3 Sections of trajectories of inversion of one amino group of urea in the gas phase. | |
Table 4 Frequencies of normal modes of urea in the gas phasea, cm−1
| Mode | Exp.b | B3LYP/6-31G* | U1 | U2 | U3 |
|---|
| Unscaled, harmonic approximation. Assignment of King.32 Assignment is difficult. |
|---|
| ν(CO) | 1734 | 1840 | 1723 | 1723 | 1731 |
| δas(NH2) | 1594 | 1663 | 1446 | 1453 | 1449 |
| δs(NH2) | 1594 | 1660 | 1586 | 1581 | 1585 |
| νas(CN) | 1394 | 1427 | 1702 | 1716 | 1698 |
| ρs(NH2) | | 1198 | 916 | 895 | 1027 |
| ρas(NH2) | 1014 | 1065 | 819 | 775 | 956 |
| νs(CN) | 960 | 955 | 815 | 804 | 830 |
| ω(CO) | 790 (?) | 789 | 817 | 776 | 793 |
| ωs(NH2)c | 227 (?) | 631 | 673 | 124 | 235 |
| ωas(NH2)c | | 588 | 568 | 376 | 440 |
| δ(CO)c | 578 (?) | 557 | 505 | 534 | 519 |
| δ(CN) | 410 (?) | 473 | 456 | 453 | 461 |
| τas(NH2)c | 618 (?) | 457 | 646 | 585 | 583 |
| τs(NH2)c | | 395 | 550 | 550 | 581 |
Table 5 Characteristics of N-inversion from trajectories
| | Gas phase | Aqueous solution |
|---|
| U1 | U2 | U3 | U1 | U2 | U3 |
|---|
| Mean life time of a pyramidal amino group. Averaged amplitude. |
|---|
| ν/cm−1 | 645 | 211 | 172 | 506 | 179 | 118 |
| τ/psa | 0.026 | 0.080 | 0.097 | 0.033 | 0.094 | 0.145 |
| yav/Åb | 0.095 | 0.231 | 0.236 | 0.161 | 0.301 | 0.330 |
Altogether, U1 is defined to be rigid and planar. This is probably the model used in previous simulations of urea–water mixtures. U2 defines a slow periodic high-amplitude mode that describes coupled N-inversion as the first band in the vibrational spectrum. Both models define N-inversion as fundamental vibration, so its frequency does not depend on temperature. In contrast to that, U3 defines an activation barrier and thus a temperature-dependent rate of N-inversion. The frequency counted at room temperature and the amplitudes are near similar to those in U2.
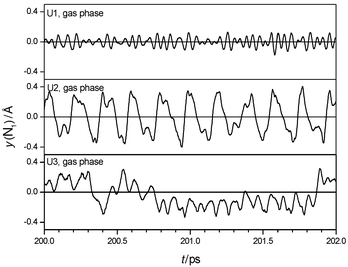
The calculated IR spectrum of U1 surrounded by water is shown in Fig. 2. Spectra for the other models are similar. The spectra are dominated by intermolecular motions of the solute in a solvent cage below 300 cm−1, and changes of the molecular dipole perpendicular to the CON2 plane between 400 and 1000 cm−1. Trajectories of N-inversion are shown in Fig. 4, the counted frequencies are summarized in Table 5. The trajectories reveal that the two amino groups invert independently in solution. Inversion amplitudes are increased compared to the gas phase, and counted frequencies of all models are red-shifted, corresponding to increased averaged lifetimes of pyramidal states. Observations like these are similar to those reported in ref. 49 from classical MD with coupled quantum mechanical/empirical
potentials of a formamide–water system. In addition, inversion events become more aperiodic in time, so calculated IR-spectra show only a weak and broad band at about 900 cm−1, which probably corresponds to the ω(CO) vibration. The inversion frequency is not detected in the IR spectra or the Fourier transforms of the trajectories. In summary, the general vibrational characteristics regarding planarity, floppiness and scaled inversion behaviour of all three models is retained after hydration, compared to the gas phase. The solvent affects amplitudes, frequencies and periodicity of inversion events.
 |
| | Fig. 4 Sections of trajectories of inversion of one amino group of urea in aqueous solution. | |
3.2 Structure of the hydrate shell
The atom-centred radial distribution functions (RDF's, see Fig. 5) indicate the presence of a well defined hydrate shell of urea up to a distance of 5.5 Å from its centre of mass. Analyses of the RDF's by means of a nearest neighbour approach50
(see Fig. 6 for the carbonyl oxygen) show that subsequent peaks observed for all atoms except the carbonyl carbon are due to water molecules coordinated directly to other solute atoms. Only the C–OW RDF (see Fig. 5) indicates the presence of a weak second hydrate shell from 7 to 9 Å. Bulk water is observed beyond 12 Å from urea. The coordination numbers of urea atoms computed over the first X–HW
(X = O, N) or X–OW
(X = H, C) peaks are summarized in Table 6.
They reveal every solute atom except C to be coordinated with approximately one (N, H) or two (O) distinct water molecules, yielding an idealized number of 8 water molecules directly coordinated to urea. The complete hydrate shell contains additional water molecules to fill the gaps between the directly coordinated solvent molecules. The exact time-averaged numbers of directly coordinated water are lower than these ideal numbers due to the fluidity of the hydrate shell. They range from 5.4 (U1) to 6.2 (U3), mainly caused by the different coordination numbers of the N atoms. This difference becomes also apparent from the first peak of the N–HW RDF's (see Fig. 5). As expected, pyramidal amino groups with certain lifetimes cause an increased probability of hydrogen bonds to a nitrogen atom. The planar model U1 shows H-bonds from water to nitrogen to some extent, but no sharp peak of the RDF is
obtained. These observations are not made in other simulations of aqueous urea solutions. Here the first peak of the N–OW RDF occurs at about 3 Å similar to our simulations, but no N–HW peak is present due to the restriction to planar urea and parameterization without taking the possibility of N–HW hydrogen bonds into account. Thus, the coordination numbers of urea reported previously are smaller (4 to 5.7). From caloric measurements it is argued that about 5 water molecules are directly coordinated to urea.51 Recent measurements of diffusion coefficients in urea–water mixtures by NMR52 point to an average hydration number of 6.7. Our results agree with these approximate numbers.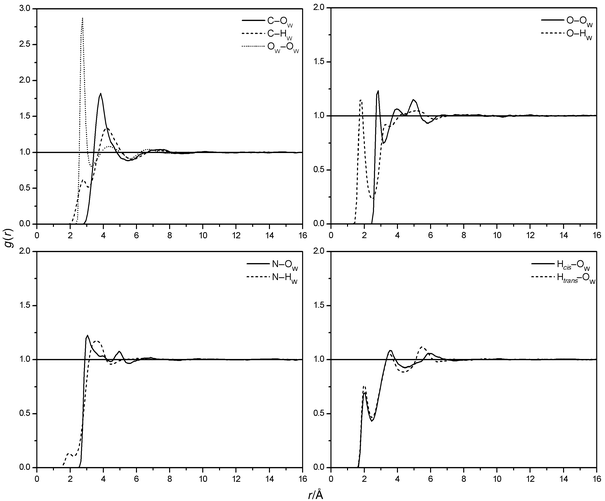 |
| | Fig. 5 Urea–water and water–water RDF's (U1). | |
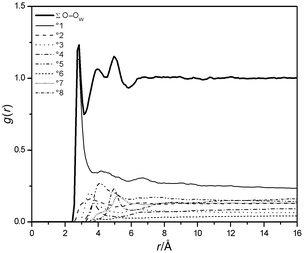 |
| | Fig. 6 Contributions to the O–OW RDF from the °xth neighbours of a water molecule (U1). | |
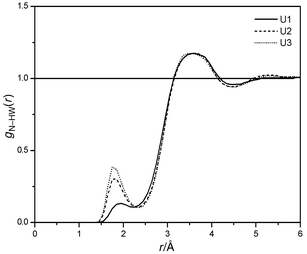 |
| | Fig. 7 N–HW RDF's of U1, U2 and U3. | |
Table 6 Coordination numbers of urea
| | rcut/Å | U1 | U2 | U3 |
|---|
| Sum of directly coordinated water molecules. |
|---|
| C–OW | 5.5 | 20.78 | 20.65 | 20.61 |
| O–HW | 2.5 | 1.937 | 1.778 | 1.828 |
| N–HW | 2.3 | 0.255 | 0.470 | 0.558 |
| Hcis–OW | 2.5 | 0.780 | 0.782 | 0.809 |
| Htrans–OW | 2.5 | 0.852 | 0.889 | 0.884 |
| Σa | | 5.711 | 6.060 | 6.330 |
The regions of the hydrate shell influenced by nitrogen inversion can be identified by the solute–solvent pair correlation function in a 3D-grid around the solute in a solute-centered coordinate system. Resulting iso-probability surfaces of solvent atom positions are shown in Fig. 8 for model U1 and U3. Model U2 has intermediate characteristics. The surfaces represent the peaks of the RDF's resolved in three-dimensional space, which are cut at a certain value. From these plots, the time-averaged positions and orientations of directly coordinated and also of some remaining solvent molecules can be identified. It becomes apparent that the overall hydrate shell is nearly spherical. The water molecules connected to the carbonyl oxygen (ideally two) are found in a defined distance range, but their angular distribution is rather broad. This observation is probably the result of the low atomic charge assigned to oxygen, and of the vibrations of
the NH2 groups with coordinated water. This feature was reported in nearly all previous MD simulations. The solvent molecules coordinated with urea hydrogens are located mostly in the urea plane. Like the ones connected to the carbonyl oxygen, they show a more or less broad distribution over both sides, depending on the flexibility of the solute. The peaks of the N–HW and N–OW RDF's (see Fig. 5) split into four regions in space. They are the result of averaging the hydrate shells of the four stereoisomers of urea in time (E and Z conformation, ideally C2 and Cs symmetry), each forming ideally one N–HW hydrogen bond per nitrogen. Interestingly, U1 shows a certain probability of finding water oxygens in this region, but fewer water hydrogens. So the position of water molecules in a region suitable for hydrogen bonds to N
seems to be predetermined in U1, probably as result of the water network around urea. However, H-bonds to N can only be formed in the case of slow large-amplitude N-inversions in the case of U2 and U3. In other words, the scaled parameterization of nitrogen inversion of urea presented in this paper mainly influences the rotational dynamics of water molecules in the first hydrate shell of urea in regions perpendicular of the carbonyl plane.
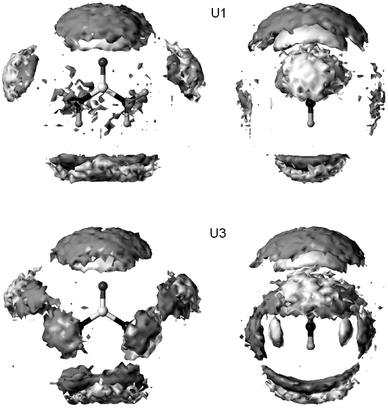 |
| | Fig. 8 Iso-probability surfaces of solvent atom positions around urea in orthographic view (dark gray: OW, light gray: HW). | |
3.3 Local perturbations of water structure
The size of the systems presented in this work allows for a detailed inspection of water properties beyond the first hydrate shell and comparison with bulk water. As shown in Fig. 9, water density is perturbed by urea up to 10 Å. This behaviour is already deduced from the RDF (see Fig. 5). The dominating contribution to the excess running coordination number plotted in Fig. 9 originates from the size of urea compared with a water molecule. However, this effect is nearly compensated up to 4.5 Å. This region covers the first and second hydrate shell of a SPC/E water molecule, and the first hydrate shell of urea (see Fig. 5). Because of this match, the small value of ΔNXS
≈
−1 calculated for the whole simulation boxes is already reached here. It has to be mentioned, that the particular value of ΔNXS
depends strongly on the water potential. It is known that all empirical water models yield a first peak of the water–water RDF that is too high compared with neutron diffraction data.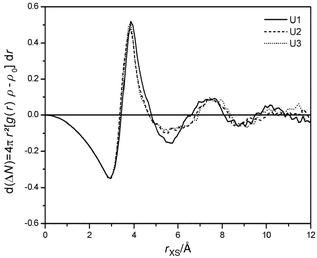 |
| | Fig. 9 Excess running coordination numbers of urea. | |
A second local perturbation of water density follows up to 8 Å, corresponding to the first valley of the urea–water RDF and the second valley of the water–water RDF. Two reasons for reduced relative water density (valleys in RDF's) can be given. First, strongly coordinated water molecules in the first shell cause a lower probability of randomly distributed solvent behind them because of their reduced mobility relative to the bulk solvent. Second, mobile members of the first shell also enhance the mobility of water at larger distances. As shown in Fig. 10, both cases are found. There exist density gaps in water due to solute molecules that are fixed at urea oxygen and the hydrogens, but also perpendicular to the molecular plane behind water molecules that could form hydrogen bonds to nitrogen. The resulting effect of density reduction is the largest for the planar quickly vibrating model U1 (corresponding to the largest negative
value of ΔNXS
(4.5 Å < R < 7 Å, see Fig. 9), whereas the other models approach the behaviour of pure water.
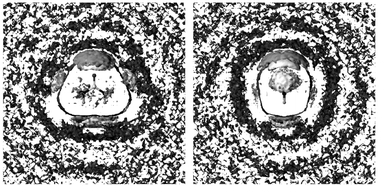 |
| | Fig. 10 Urea–water PDF's of U1 in orthographic view (same as Fig. 8) and regions with reduced water density (dark gray). | |
The evaluation of a measure of the hydrogen bond network of water yields a consistent interpretation. We measure the extent of this network by the number nHB/nOc, where nHB is the number of hydrogen bonds per water molecule to other solvent molecules, and nOc is its coordination number with surrounding water oxygen. We applied distance and angle criteria for the definition of hydrogen bonds and coordination numbers (rOO < 3.25 Å, rOH < 2.45 Å, θOOH < 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °). The quotient of these two numbers represents the probability of formation of a hydrogen bonded network in water. A number of 1 means that all neighbours of a water molecule form H-bonds to it. A lower number indicates network defects by an increasing amount of five- or six-coordinated water, or by a reduced number of H-bonds. The first possibility
yields an increased density similar to melting of ice, the second is consistent with a reduced water density similar to heating of liquid water at ambient temperature. Typical values of nHB/nOc for water from MD simulations using empirical potentials amount to about 0.8–0.9 depending on the criteria of H-bond definition. As shown in Fig. 11, the structure of water measured by nHB/nOc is perturbed up to about 10 Å from urea. The largest effect is identified from 4 to near 6 Å as an increased structure of water. This is the region of water molecules that are coordinated and hydrogen bonded to molecules directly coordinated to urea. The following reduced network structure is present up to 7 Å. Since water density is reduced here, it is caused by increased water mobility in this region. Differences between the three urea models are hard to detect. The
only visible effect is seen around 4 Å from urea. U1 yields more water–water H-bonds than the other two models here. This finding can be interpreted as “hydrophobic solvation” of U1. It is probably caused by the coordination of those water molecules with surrounding water that form a N-HW H-bond in U2 and U3.
°). The quotient of these two numbers represents the probability of formation of a hydrogen bonded network in water. A number of 1 means that all neighbours of a water molecule form H-bonds to it. A lower number indicates network defects by an increasing amount of five- or six-coordinated water, or by a reduced number of H-bonds. The first possibility
yields an increased density similar to melting of ice, the second is consistent with a reduced water density similar to heating of liquid water at ambient temperature. Typical values of nHB/nOc for water from MD simulations using empirical potentials amount to about 0.8–0.9 depending on the criteria of H-bond definition. As shown in Fig. 11, the structure of water measured by nHB/nOc is perturbed up to about 10 Å from urea. The largest effect is identified from 4 to near 6 Å as an increased structure of water. This is the region of water molecules that are coordinated and hydrogen bonded to molecules directly coordinated to urea. The following reduced network structure is present up to 7 Å. Since water density is reduced here, it is caused by increased water mobility in this region. Differences between the three urea models are hard to detect. The
only visible effect is seen around 4 Å from urea. U1 yields more water–water H-bonds than the other two models here. This finding can be interpreted as “hydrophobic solvation” of U1. It is probably caused by the coordination of those water molecules with surrounding water that form a N-HW H-bond in U2 and U3.
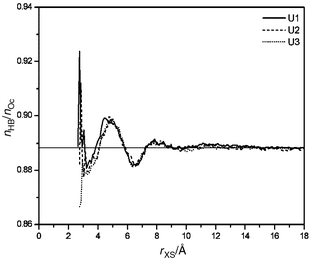 |
| | Fig. 11 Perturbation of the H-bond network of water in shells at increasing distance from urea. | |
The statements given above are also verified by calculating the translational diffusion coefficient of water in different regions of the solution. We obtained the diffusion coefficients by integration of the velocity autocorrelation function of the centre of mass of water molecules that are found in a defined distance range from urea for the whole correlation time of 3 ps. This approach is necessary in order to assign a diffusion coefficient of moving particles to a location. Since the number of water molecules that fulfil this criterion is very small, the obtained diffusion coefficients bear a high amount of error. So differences between the three models are statistically not significant. However, the result of D
= 2.9 (0–4.5 Å), 3.25 (4.5–7 Å), 3.05 (7–12 Å), 3.08 × 10−5 cm2 s−1
(12–18 Å) reveals that translational mobility of water
is indeed enhanced over the bulk value behind the first hydrate shell.
3.4 Solvation thermodynamics
The contributions to the hydration enthalpy of urea obtained with all three urea models are given in Table 7. Although the models are defined to have the same parameters for non-bonding interactions, there exist significant differences of the total urea-water interaction energies EXS. The slow vibrating model U3 shows the tightest solvent–solute interactions, followed by U2 and U1 as expected. When comparing the solute binding energies EXS with results previously reported from additive potentials with planar urea models, we mostly find smaller absolute amounts [−15.4 kcal mol−1,15
−21.0 kcal mol−1,10
−31.3 kcal mol−1 (ref. 9)] because of the lower permanent dipole moment of urea used (about 4 D, which is too small compared
with experiment).
Table 7 Thermodynamic properties of aqueous urea solutions, kcal mol−1
| | U1 | U2 | U3 |
|---|
| Defined by eqn. (5). |
|---|
| EXS | −35.6 ± 0.1 | −38.1 ± 0.2 | −40.8 ± 0.3 |
| ΔES | 25.9 ± 6 | 18.5 ± 5 | 25.5 ± 5 |
| ΔEX | 1.9 ± 0.1 | 2.6 ± 0.1 | 3.0 ± 0.1 |
| ΔHhyda | −7.2 ± 6 | −16.4 ± 5 | −11.7 ± 5 |
This quantity is partly compensated by the perturbation of the solvent self energy ES. Due to the statistical error no reliable comparison of the three models can be given for the overall value, which amounts to more than the half of ΔEXS, so that the hydration enthalpies obtained from our simulations are close to the experimental value of −17.34 kcal mol−1
(obtained from the standard heat of sublimation of solid urea53 and the standard heat of dissolving solid urea in water54) in the case of U2 and U3. The calculated hydration enthalpy of U1 seems to be too small because of weak solute–solvent interactions caused by the lack of N–HW hydrogen bonds. As shown by eqn. (4), ΔES can be divided into endothermic and exothermic contributions at selected distances
from the solute. Fig. 12 shows the differential contributions to ΔES. The first endothermic perturbation of the solvent self energy arises at small distances from urea as a result of excluded water, but it is nearly compensated when integration is performed over the first hydrate shell up to about 4.5 Å. The main contribution to ΔES
≈ 20 kcal mol−1 found for the whole simulation boxes is located here. The interpretation of the following smaller endothermic perturbation up to 7 Å is consistent with that given for water structure. Water becomes locally melted. This effect is the largest for the rigid model U1.
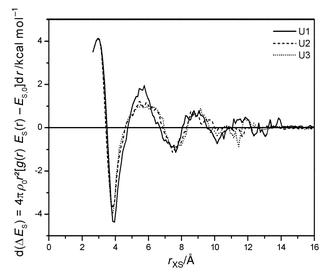 |
| | Fig. 12 Perturbation of ES in shells at increasing distance from urea. | |
4 Summary
We presented three different parameter sets of the intramolecular degrees of freedom of urea and performed classical MD of its aqueous solution with an empirical potential of almost the simplest form. Despite this crude approximation, the analysis of perturbation of water structure by means of comparison of local properties of the urea–water solution with those in pure water agrees with interpretations of previously reported simulations in terms of a general water-like behaviour of urea. Our analyses revealed that the extent and structure of its first hydrate shell is responsible for this behaviour, since it looks like the second hydrate shell of a water molecule. In addition it was shown, that urea acts the more water-like, if the possibility of N–HW hydrogen bonds for about 100 fs is taken into account. Otherwise an increased melting of water beyond the first hydrate shell is observed, which corresponds to reduced water density and enhanced solvent mobility. The
perturbation is found to range up to 7 Å from the centre of mass of urea. These findings are important when comparing urea–water mixtures with aqueous solutions of other solutes in a quantitative manner, and especially when discussing the ability of urea to preserve the structural integrity of water when hydrophobic solutes are introduced.When using experimental data of urea–water solutions as a measure, then urea models that define slow large-amplitude inversions of amino groups with frequencies of 100–200 cm−1 are more appropriate to describe this system than using amide-like potentials. Regarding the mechanism of inversion it was found that the definition of a slow fundamental vibration of planar urea has the same effect on its hydrate shell as the introduction of a kinetic barrier of inversion into a C2 structure, when additive potentials are used for classical MD.
References
- H. S. Frank and F. Franks, J. Chem. Phys., 1968, 48, 4746 CrossRef CAS.
- R. H. Stokes, Aust. J. Chem., 1967, 20, 2087 CAS.
- J. A. Schellman, C. R. Trav. Lab. Carlsberg, Ser. Chim., 1955, 29, 233 Search PubMed.
- G. C. Kreschek and H. A. Scheraga, J. Phys. Chem., 1965, 69, 1704 CrossRef.
- R. A. Kuharski and P. J. Rossky, J. Am. Chem. Soc., 1984, 106, 5794 CrossRef.
- R. A. Kuharski and P. J. Rossky, J. Am. Chem. Soc., 1984, 106, 5786 CrossRef.
- P. O. Åstrand, A. Wallqvist, G. Karlström and P. Linse, J. Chem. Phys., 1991, 95, 8419 CrossRef.
- E. S. Boek and W. J. Briels, J. Chem. Phys., 1993, 98, 1422 CrossRef CAS.
- J. Hernández-Cobos, I. Ortega-Blake, M. Bonilla-Marín and M. Moreno-Bello, J. Chem. Phys., 1993, 99, 9122 CrossRef CAS.
- P. O. Åstrand, A. Wallqvist and G. Karlström, J. Phys. Chem., 1994, 98, 8224 CrossRef.
- J. Pranata, J. Phys. Chem., 1995, 99, 4855 CrossRef CAS.
- J. Tsai, M. Gerstein and M. Levitt, J. Chem. Phys., 1996, 104, 9417 CrossRef CAS.
- F. S. Nandel, R. Verma, B. Singh and D. V. S. Jain, Pure Appl.
Chem., 1998, 70, 659 CAS.
- A. Wallqvist, D. G. Covell and D. Thirumalia, J. Am. Chem. Soc., 1998, 120, 427 CrossRef CAS.
- A. Tovchigrechko, M. Rodnikova and J. Barthel, J. Mol. Liq., 1999, 79, 187 CrossRef CAS.
- A. Idrissi, F. Sokolic and A. Perera, J. Chem. Phys., 2000, 112, 9479 CrossRef CAS.
- K. A. Sharp, B. Madan, E. Manas and J. M. Vanderkooi, J. Chem. Phys., 2001, 114, 1791 CrossRef CAS.
- H. Guth, G. Heger, S. Klein, W. Treutmann and C. Scheringer, Z. Kristallogr., 1980, 153, 237 Search PubMed.
- J. E. Worsham, H. A. Levry and S. W. Peterson, Acta Crystallogr., 1957, 10, 319 CrossRef.
- A. W. Pryor and P. L. Sanger, Acta Crystallogr., Sect A., 1970, A26, 543.
- S. Swaminathan, B. M. Craven and R. K. McMullan, Acta Crystallogr., Sect. B, 1984, B40, 300 CrossRef.
- R. D. Brown, P. D. Godfrey and J. Storey, J. Mol. Spectrosc., 1975, 58, 445 CrossRef CAS.
- P. D. Godfrey, R. D. Brown and A. N. Hunter, J. Mol. Struct., 1997, 413, 405 CrossRef.
- M. Kontoyianni and P. J. Bowen, J. Comp. Chem., 1992, 13, 657 Search PubMed.
- D. A. Dixon and N. Matsuzawa, J. Phys. Chem., 1994, 98, 3967 CrossRef CAS.
- B. Kallies and R. Mitzner, J. Mol. Struct. (THEOCHEM), 1998, 428, 267 CrossRef CAS.
- T. Strassner, J. Mol. Model., 1996, 2, 217 CrossRef CAS.
- B. Kallies and R. Mitzner, J. Mol.
Model., 1998, 4, 183 CrossRef CAS.
- C. Lee, E. A. Stahlberg and G. Fitzgerald, J. Phys. Chem., 1995, 99, 17737 CAS.
- G. E. Walrafen, Water: A Comprehensive Treatise, Plenum, New York, 1972, vol. 1.
- J. Martí, J. A. Padro and E. Guàrdia, J. Chem. Phys., 1996, 105, 639 CrossRef CAS.
- S. T. King, Spectrochim. Acta, Part A, 1972, 28, 165 CrossRef CAS.
- R. Keuleers, H. O. Desseyn, B. Rousseau and C. Van Alsenoy, J. Phys. Chem. A, 1999, 103, 4621 CrossRef CAS.
- M. Spoliti, A. Pieretti, L. Bencivenni and N. Sanna, Electron. J. Theor. Chem., 1997, 2, 149 Search PubMed.
- A. Vijay and D. N. Sathyanarayana, J. Mol. Struct., 1993, 295, 245 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269 CrossRef CAS.
- R. W. Hockney, Methods Comput. Phys., 1970, 9, 136 Search PubMed.
- W. D. Kumler and G. M. Fohlen, J. Am. Chem. Soc., 1942, 64, 1944 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R.
Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian98 A5, Gaussian, Inc., Pittsburgh, PA, 1998.
- D. A. Case, D. A. Pearlman, J. W. Caldwell, T. E. Cheatham III, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, D. M. Ferguson, R. J. Radmer, G. L. Seibel, U. C. Singh, P. K. Weiner and P. A. Kollman, AMBER 5.0.1, UCSF, 1997.
- SYBYL 6.6, Tripos, Inc., St. Louis,
MO, 1999.
- R. Friedberg and J. E. Cameron, J. Chem. Phys., 1970, 52, 6049 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Claredon Press, Oxford, 1987 Search PubMed.
- A. A. Chiavlo, P. T. Cummings, J. M. Simonson and R. E. Mesmer, J. Chem. Phys., 1999, 110, 1064 CrossRef.
- B. D. Bursulaya and H. J. Kim, J. Chem. Phys., 1998, 109, 4911 CrossRef CAS.
- S. Chalmet and M. F. Ruiz-López, J. Chem. Phys., 1999, 111, 1117 CrossRef CAS.
- P. K. Mehrotra and D. L. Beveridge, J. Am. Chem. Soc., 1980, 102, 4287 CrossRef CAS.
- J. N. Spencer and J. W. Hovick, Can. J. Chem., 1988, 66, 562 CAS.
- L. Costantino, G. D’Errico, O. Ortona and V. Vitagliano, J. Mol. Liq., 2000, 84, 179 CrossRef CAS.
- K. Suzuki, S. Onishi, T. Koide and S. Seki, Bull. Chem. Soc. Jpn., 1956, 29, 127 CAS.
- E. P. Egan and B. B. Luff, J. Chem. Eng. Data, 1966, 11, 192 CrossRef CAS.
Footnote |
| † 1 D (debye) ≈ 3.33564 × 10−30 C m. |
|
| This journal is © the Owner Societies 2002 |
Click here to see how this site uses Cookies. View our privacy policy here.