The siting of Cu(II) in mordenite: a theoretical spectroscopic study
Annelies
Delabie
*a,
Kristine
Pierloot
a,
Marijke H.
Groothaert
b,
Bert M.
Weckhuysen
b and
Robert A.
Schoonheydt
b
aDepartment of Chemistry, University of Leuven, Celestijnenlaan 200F, B-3001 Heverlee-Leuven, Belgium. E-mail: annelies.delabie@chem.kuleuven.ac.be
bCenter for Surface Chemistry and Catalysis, University of Leuven, Kasteelpark Arenberg 23, B-3001 Heverlee-Leuven, Belgium
First published on 18th December 2001
Abstract
The siting of Cu(II) in mordenite has been studied by ab initio calculations on large cluster models, representing the cation exchange sites in mordenite. Partial geometry optimizations, based on density functional theory (DFT), were performed to obtain the structure of the coordination environment of Cu(II) at the different sites. The ligand field spectra and EPR g-tensors of these clusters were then calculated by means of multiconfigurational perturbation theory (CASPT2). The calculated results were compared with experimental information, obtained by diffuse reflectance spectroscopy (DRS) and EPR. The calculations indicate that at low exchange levels Cu(II) is coordinated to oxygen six-rings in the main channel of mordenite, in the presence of two aluminiums. At higher loadings, six- or five-rings containing only one aluminium also become occupied, where Cu(II) is coordinated as a single ion, not as (Cu–OH)+. The calculations indicate also that in fully dehydrated mordenite, the twisted eight-ring (site A) is not occupied by Cu(II).
1 Introduction
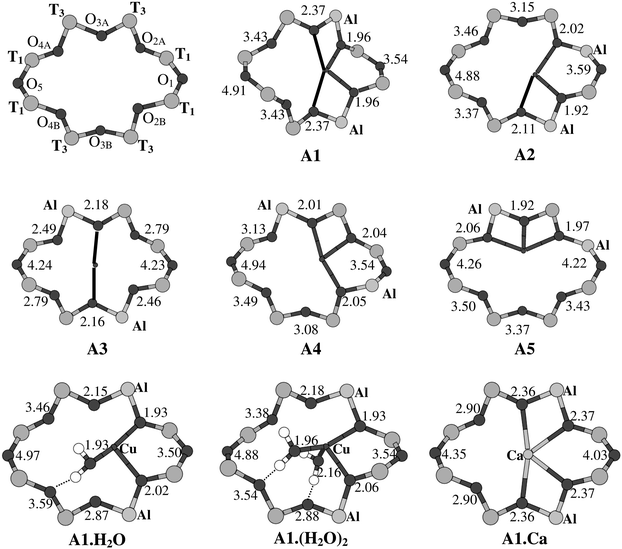
Zeolites containing transition metal ions are widely studied, because of their interesting adsorptive and catalytic properties.1–3 It has been found that pentasil zeolites (ZSM-5, ferrierite and mordenite) loaded with transition metal ions may display high activities towards the decomposition and reduction of NO.4–6 Despite the large amount of available experimental and theoretical studies, the exact nature of the active site and the reaction mechanism are still under debate. Detailed knowledge of the location and coordination of the transition metal ions in these zeolites is an important first step to a better understanding of these materials. Over the years, X-ray diffraction (XRD), EPR and electronic spectroscopy have proven to offer valuable information concerning the location and coordination of isolated Cu(II) ions in dehydrated zeolite matrices.According to our knowledge, an XRD structure of fully dehydrated Cu(II) exchanged mordenite is not yet available. Most of the interpretations of the siting of Cu(II) in mordenite are therefore based on the XRD structure of dehydrated Ca2+ mordenite.7 The structure of mordenite (MOR), with indication of the possible exchangeable cation sites, is shown in Fig. 1. Four sites are occupied by Ca2+. Firstly, Ca2+ ions are found in site A, a twisted eight-ring located in the elliptical eight-ring channels of mordenite. The Ca2+ ion in this site is coordinated to six lattice oxygens in C2h symmetry. A second occupied site is the six-ring in the twelve-membered ring channel (site E), where Ca2+ is coordinated to four oxygens. A smaller portion of the Ca2+ ions is present in the boat-shaped site, located in the eight-ring channel of the zeolite. This site is composed of a non-planar six-ring (site C) in between two bent five-rings (see Fig. 1). Finally, Ca2+ ions were also found in the circular eight-rings, situated in the walls of the large twelve-ring channel (site D).
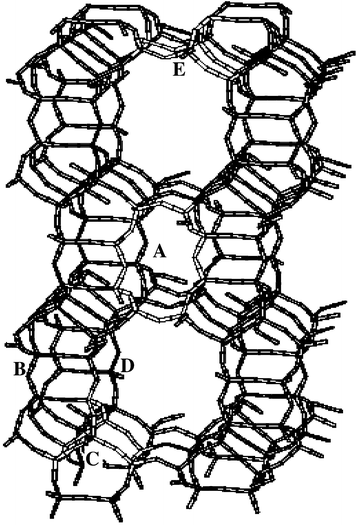 | ||
| Fig. 1 Structure of mordenite with indication of the cation sites (A, B, C, D, and E). The structure of site E, A and C are indicated in light-grey. | ||
XRD data for hydrated and partially dehydrated Cu(II) mordenite have also been reported.8 In partially dehydrated Cu(II) mordenite, three different Cu(II) cation sites are occupied. In site A, Cu(II) is not only coordinated to six lattice oxygens of the eight-ring, but also to two water molecules. The second exchange site which is occupied by Cu(II) is site E, where Cu(II) is coordinated to four lattice oxygens. The last and least occupied site is site D (see Fig. 1), where Cu(II) is bound to three oxygens.
Spectroscopic techniques like EPR and DRS have also been applied succesfully to investigate the coordination of Cu(II) in mordenite. The interpretation of the information obtained from such experiments is however not always straightforward, and has led to different proposals in the past. In the first EPR study of Cu(II) exchanged mordenite, two types of Cu(II) cations were detected, both in an axial environment.9 The two coordination environments were described as either four-coordinated square planar (characterized by gzz![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =2.27) or five-coordinated square-pyramidal (characterized by gzz=2.32).
=2.27) or five-coordinated square-pyramidal (characterized by gzz=2.32).
An interpretation of combined DRS–EPR data of Cu(II) mordenite, based on angular overlap model (AOM) calculations, has been provided.10,11 According to this interpretation only site A is occupied at low Cu(II) exchange levels, the weak coordination with six oxygens of this site giving rise to the EPR signal with a gzz value of 2.32. At higher copper loadings, a second exchange site, i.e. site E, becomes occupied. The four-fold Cu–oxygen coordination at this site was held responsible for the second EPR signal with gzz=2.27.
More recently, an alternative interpretation of the siting of Cu(II) in various zeolite matrices was provided by Wichterlová
et al.12,13 They studied the Cu(II) coordination by means of Cu+ luminescence, IR of adsorbed NO on Cu(II), and EPR. From the combined information obtained from these techniques, it was concluded that the Cu(II) siting in high silica zeolites is essentially controlled by the local Si–Al sequences in the framework: Cu(II)
(characterized by gzz=2.32) is located preferentially in the vicinity of two framework aluminiums. Here it obtains the square-pyramidal oxygen coordination corresponding to the first, gzz=2.32, EPR signal. A second type of Cu(II), characterized
by a gzz value of 2.27, has a square planar oxygen coordination and is located in the vicinity of only one aluminium. The possibility of an extra-framework oxygen ligand was, for the latter site, also considered.
In the present study, high-level theoretical calculations have been performed in order to obtain a sound interpretation of the two EPR signals of Cu(II) in mordenite. The calculations involve (partial) geometry optimizations, based on DFT, performed on large cluster models representing Cu(II) at different possible exchange sites in mordenite. The role of the aluminium distribution in the coordination and siting of Cu(II) is considered in detail. Using the DFT optimized structures, multiconfigurational perturbation theory (the CASPT2 method) is employed to calculate the ligand field spectra and EPR g-factors of the different cluster models. The calculated spectra are confronted with the experimental DRS and EPR data, and are used both for the interpretation of these spectra, and to distinguish between Cu(II) at different oxygen sites in mordenite. We note that the DRS and EPR experiments reported in this work did not reveal any new information.11 The main purpose of including the experimental results is to provide a basis for discussion of the theoretical calculations. Furthermore, our calculations are of course limited by the size of the cluster models used. Recently, a method has been developed which makes it possible to consider also the effect of the further zeolite environment by using an embedding procedure, in which the quantum mechanical description of the cluster is coupled to a classical description of the interactions between the cluster and its further environment.14–17 Nevertheless, the present limited cluster approach has already been applied successfully in previous studies on the coordination and spectroscopic properties of Co(II) and Cu(II) in the zeolites A, Y and ZK4.18–21 The present contribution will further confirm the power of our approach.
2 Experimental
A synthetic Na-mordenite from Norton, specified as Zeolon 100, was used. A 29Si MAS NMR measurement was performed on the original Zeolon 100 to determine the Si–Al orderings in the zeolite lattice and its Si/Al-ratio. The spectrum was obtained on a BRUKER AMX300 spectrometer, with a magnetic field strength of 7 T. 5000 scans were accumulated using a pulse length of 3 μs (π/4 pulse) and a recycle delay of 20 s. The sample was spun at 4.7 kHz and tetramethylsilane (TMS) was used as the external shift reference.Four NaCuMOR samples with different Cu(II) loadings were prepared by ion exchange in aqueous CuCl2·2H2O solutions (1 (g zeolite) l−1). During exchange the pH was kept slightly acidic to avoid the formation of polynuclear Cu–hydroxyl complexes. After 24 h of exchange at room temperature the NaCuMOR was washed Cl−-free and dried at 393 K. The Cu and Na contents of the four samples were determined by ICP (inductively coupled plasma), and the analytical results are given in Table 1. The subscript in the sample symbol indicates the number of Cu(II) ions per unit cell. The four samples contain 0.05, 0.09, 0.35 and 0.75 Cu(II) ions per unit cell respectively. The CuMOR samples with grain sizes between 0.25 and 0.4 mm were dehydrated at 723 K in an O2-flow to preserve all copper in the (+II) valence state. After 24 h at 723 K, complete dehydration can be assumed.22 The samples were cooled in an O2-stream and flushed with He. DRS spectra were recorded on a Varian Cary 5 UV–VIS–NIR spectrophotometer at room temperature. They were measured against a halon white reflectance standard in the range 200–2500 nm. The EPR spectra were recorded at 120 K on a Bruker ESP 300E instrument operating in X-band with a microwave power of 20 mW. The Cu(II) EPR signals were registered in the field region of 2250–3950 G. For the simulation of the EPR spectra the Simpow program23 was used.
3 Theoretical methods
3.1 Geometry optimizations
Based on the information obtained from XRD on Ca(II) and partially dehydrated Cu(II) mordenite, a series of cluster models were constructed, representing the most likely coordination sites of Cu(II) in fully dehydrated mordenite: site E, site A as a representative of an oxygen eight-ring site, and site C of the boat-shaped site in the eight-ring channel (see Fig. 1). All cluster models were terminated by dangling T–OH bonds (T=Si,Al). The starting geometry of the cluster models is based on XRD data of dehydrated Ca2+-exchanged mordenite.7 Partial geometry optimizations of these cluster compounds were performed, in which the position of all atoms was optimized, except for the dangling OH bonds. For the latter, the O positions as well as the OH orientation were kept fixed at the XRD values, and only
the OH bond distances were optimized.
Site A consists of a simple eight-ring, and the model cluster used to describe this site therefore consists of CuO8T8(OH)16. On the other hand, sites E, C are six-rings with an additional O–T–O bridge, which was also included in the calculations, thus giving a CuO8T7(OH)12 cluster. Alternatively, these sites may be seen as a combination of two five-rings, sharing three T atoms. The actual boat-shaped site is in fact composed of site C, surrounded by two strongly bent five-rings (see Fig. 1). To examine the possibility that Cu(II) might coordinate to these five-rings rather than to site C itself, geometry optimizations were also performed on the representative CuO5T5(OH)10 cluster models. However, in the final structures, Cu(II) was found to coordinate to only two oxygens of the O5T5 ring, forming instead two additional bonds to the terminating OH groups corresponding to the oxygen six-ring of site C. It was therefore concluded that Cu(II) at the boat-shaped site prefers site C over its five-ring neighbours, and the latter were not considered for the calculation of spectroscopic properties.
The mordenite used in this study has a Si/Al ratio of 5.7 (in general, the Si/Al ratio in mordenite can range from 5 to 20), therefore, cluster models without or with one or two aluminium atoms were considered. In accordance with Loewenstein's rule,24 aluminiums were never placed in adjacent tetrahedra. Furthermore, based on 29Si NMR measurements, Takaishi et al. developed an additional rule for the aluminium distribution in mordenite, stating that two aluminiums never occur together inside one five-ring.25,26 The validity of this rule was examined and confirmed for site E (by considering also clusters models for which this rule is not obeyed). For site C and its surrounding five-rings, only clusters containing a maximum of one Al per five-ring were considered.
All geometry optimizations were performed with density functional theory, using the Turbomole code.27 The B3LYP functional was used in combination with the basis sets from Schäfer et al.:28 for copper the double-zeta basis set, enhanced with diffuse p, d, and f functions was used. For oxygens inside the ring, a triple-zeta basis set with one polarization function was used, while for Si, Al, and the O, H of the dangling OH bonds double-zeta basis sets were employed. The optimizations of all clusters considered were restarted several times from a different initial position of Cu(II), to gain confidence that the final results indeed correspond to the global structural minimum. Other local minima could be located for most clusters. These local minima were always found at a significantly (more than 4 kcal mol−1) higher energy than the global minimum.
For all cluster models, a similar geometry optimization was also performed on the bare cluster in the absence of Cu(II), thus allowing for an estimation of the binding energy (BE) of Cu(II) at the different sites. The latter was calculated as follows:
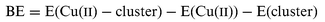 | (1) |
For one cluster (A1 in Fig. 7, see later) we have also examined whether the coordination of Cu(II) can be completed by additional water ligands. Finally, we also have looked in some more detail into the possibilities for charge compensation when Cu(II) is coordinated to an oxygen ring containing only one aluminium. It has been suggested12,29–31 that Cu(II) coordinated in the proximity of only one Al should carry an OH− group as a fifth ligand, to compensate for the local surplus of positive charge. To check this surmise, we performed additional calculations on two clusters with one Al, placed as in clusters E1 and C1 (represented in Fig. 5 and 6 respectively, see later), but now containing an extra OH− group coordinated to Cu(II). Geometry optimizations were performed with the same procedure as described above. Triple-zeta basis sets with one polarization function were used for the atoms of the adsorbed ligands.32
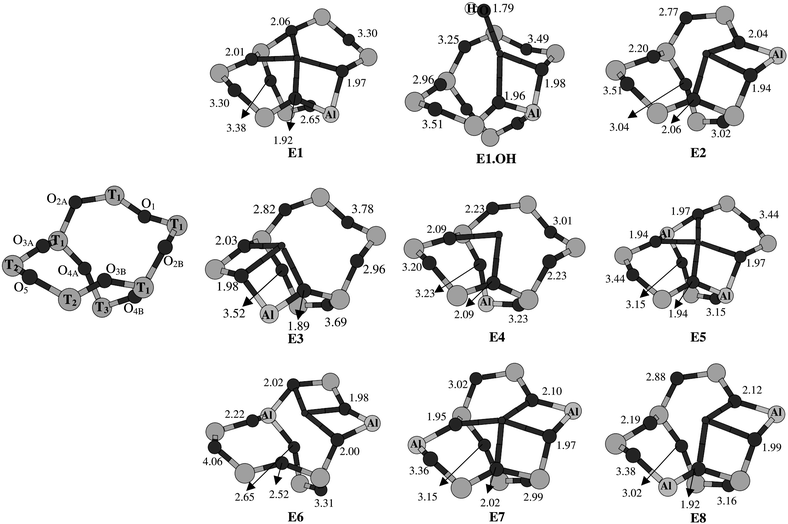 | ||
| Fig. 5 Labeling of the O- and T-atoms in site E, and coordination environments of Cu2+ in site E (clusters E1–E8, E1.OH). Cu–O distances are given in Å. | ||
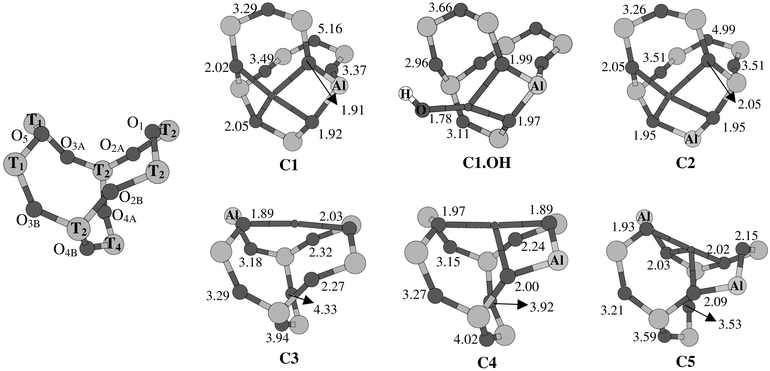 | ||
| Fig. 6 Labeling of the O- and T-atoms in site C, and coordination environments of Cu2+ in site C (clusters C1–C5, C1.OH). Cu–O distances are given in Å. | ||
 | ||
| Fig. 7 Labeling of the O- and T-atoms in site A, and coordination environments of Cu2+ (clusters A1–A5) and Ca2+ (cluster A1.Ca) in site A. Cu–O or Ca–O distances are given in Å. In clusters Al·(H2O) and Al·(H2O)2, one and two additional water molecules are adsorbed on Cu(II). | ||
3.2 Calculation of spectroscopic features
To reduce computational costs (being much heavier for an ab initio CASSCF/CASPT2 than for a B3LYP-DFT calculation), slightly smaller cluster models were constructed for the calculation of spectroscopic properties. Starting from the optimized clusters, all dangling OH groups were replaced by H (T–H bond distances were reoptimized). Furthermore, in those cluster models representing sites E, C where Cu(II) was found to coordinate exclusively to lattice oxygens belonging to either the six-ring or one of the five-rings (the distance to all other oxygens being larger than 3 Å), the calculations were performed using only the respective rings, again terminated by hydrogens. The first approximation (replace T–OH by T–H) was already tested in a previous study,19 where it was found to affect the ligand field excitation energies by no more than a few hundred cm−1. The second approximation is tested here for one of the site E cluster models containing two aluminiums, positioned such that Cu(II) is coordinated in the six-ring and Cs symmetry is retained (see cluster E5 in Fig. 5, see later). The results are shown in Table 2 (first two columns). As one can see, small (<250 cm−1) differences are found between the CASPT2 excitation energies obtained with the original (including the bridge) and truncated (only the six-ring) cluster.| With bridge | Without bridge | ||||
|---|---|---|---|---|---|
| Cu[6s4p3d1f] | Cu[6s4p3d1f] | Cu[6s4p3d] | Cu[6s4p3d1f] | Cu[6s4p3d] | |
| O[3s2p1d] | O[3s2p1d] | O[3s2p1d] | O[3s2p] | O[3s2p] | |
| 2A″ | 0 | 0 | 0 | 0 | 0 |
| 2A′ | 12767 |
12719 |
12444 |
12767 |
12465 |
| 2A′ | 14515 |
14418 |
14278 |
14714 |
14538 |
| 2A″ | 14544 |
14430 |
14195 |
14615 |
14344 |
| 2A′ | 15897 |
15658 |
15521 |
15938 |
15757 |
Calculations of the electronic spectra and EPR g-tensor of the cluster models were performed by means of multiconfigurational perturbation theory based on a CASSCF wavefunction, i.e. the CASPT2 method33 using the MOLCAS-4 software.34 The basis sets employed are atomic natural orbitals (ANO),35 contracted as follows: Cu [6s4p3d1f], O [3s2p1d], Si, Al [4s3p], H [2s]. These basis sets were used for all calculations, except for a few site E models (i.e. E1, E6 in Fig. 5, see later) containing a short Cu–O distance (2.65 Å) to one of the oxygens in the O–T–O bridge, which could therefore not be removed. In this case, the Cu [6s4p3d] and the [3s2p] basis set for O were used instead. The effect of this basis set reduction was again tested for cluster E5 (Fig. 5). The results are included in Table 2. Small deviations are obtained between the CASPT2 excitation energies calculated with different combinations of basis sets, thus showing that the errors caused by the lack of polarization functions in the basis sets are similar for the ground and excited states, and therefore of little concern for the spectroscopic properties considered in this work.
The CASSCF wavefunction was obtained by distributing 11 electrons in an active space of 11 orbitals, consisting of the Cu 3d orbitals, a correlating 4d shell and the bonding O 2p orbital, corresponding with the Cu 3d orbital which is singly occupied in the ground state. CASPT2 calculations were based on state-averaged CASSCF calculations over the five doublet states, and all core orbitals were kept frozen in the CASPT2 step. A level shift of 0.3 eV, together with a back-correction technique36 was used in all CASPT2 calculations. The present CASSCF/CASPT2 approach is the same as used previously in ref. 18 and 21, where more details are given.
The effect of spin–orbit coupling was introduced by means of an effective one-electron operator:37
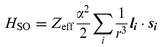 | (2) |
=17.1 (in atomic units of charge) was obtained from test calculations on the 2D ground state of Cu(II), where the effective charge was scaled until the experimental spin–orbit splitting was reproduced. The spin–orbit matrix was constructed using the CASSCF wavefunctions of the five ligand field states under consideration. However, before diagonalization the diagonal matrix elements were substituted by the more accurate CASPT2 energies. The g-tensors were calculated using second-order perturbation theory, introducing both spin–orbit coupling and the magnetic
field as simultaneous perturbations. This leads to the following expression for the tensor elements gμν
(with ge the free electron g-value):38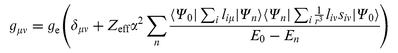 | (3) |
4 Results
4.1 Experimental results
Fig. 2 shows the 29Si MAS NMR spectrum of the Zeolon 100 MOR. The spectrum displays three broad signals for Si(2Al) (![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) Si linked to two aluminiums), Si(1Al) and Si(0Al) at about −98, −105 and −112 ppm respectively.39,40 The relative peak intensities show that 6% of the Si-atoms are linked to two aluminiums, 58% are linked to 1 Al and 36% has no neighbouring Al. With neglect of the contribution of silanols to the Si(1Al) peak the Si/Al-ratio of the mordenite lattice was calculated to be 5.7. This means that there are approximately 7 aluminiums and 41 silicons in one mordenite unit cell. Hence each unit cell contains on average 2.5 Al–O–Si–O–Al linkages.
Si linked to two aluminiums), Si(1Al) and Si(0Al) at about −98, −105 and −112 ppm respectively.39,40 The relative peak intensities show that 6% of the Si-atoms are linked to two aluminiums, 58% are linked to 1 Al and 36% has no neighbouring Al. With neglect of the contribution of silanols to the Si(1Al) peak the Si/Al-ratio of the mordenite lattice was calculated to be 5.7. This means that there are approximately 7 aluminiums and 41 silicons in one mordenite unit cell. Hence each unit cell contains on average 2.5 Al–O–Si–O–Al linkages.
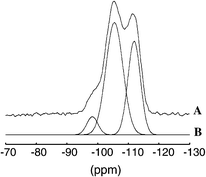
Fig. 3 shows the DRS spectra of the four dehydrated Cu(II) MOR samples in the wavenumber region 4000–20000 cm−1. The broad band in the visible with maximum intensity at about 13800 cm−1 is determined by the electronic transitions of Cu(II). When the Cu(II) loading is increased from 0.05 to 0.75 ions per unit cell, the d–d absorption band gains in intensity and broadens in both directions. In contrast to the spectra of Cu(II) in zeolite A and Y,18,41 we cannot distinguish the bands of the individual transitions. The comparison between theoretical and experimental spectra will therefore only be based on the frequency range: transitions between 8000 and 18000 cm−1 are observed experimentally. In Fig. 4, the EPR spectra of
all four Cu(II) MOR samples are shown together with the simulated spectra of Cu0.05MOR and Cu0.75MOR. The EPR spectra of Cu0.05MOR and Cu0.09MOR both consist of one Cu(II) signal, which is almost perfectly axial and corresponds to the following set of simulated parameters g(1)zz=2.33, A(1)zz=169×10−4 cm−1, g(1)xx=g(1)yy=2.07, A(1)xx=9×10−4 cm−1 and A(1)yy=22×10−4
cm−1. In the spectra of Cu0.35MOR and Cu0.75MOR a second Cu(II) signal becomes apparent, again close to axially symmetric but characterized by a smaller gzz-value. For this Cu(II) signal, present only at the higher loadings, the simulated parameters are g(2)zz=2.27, A(2)zz=191×10−4 cm−1, g(2)xx=g(2)yy=2.09, A(2)xx=18×10−4 cm−1 and A(2)yy=42×10−4
cm−1. Similar EPR spectra were reported previously.9–11
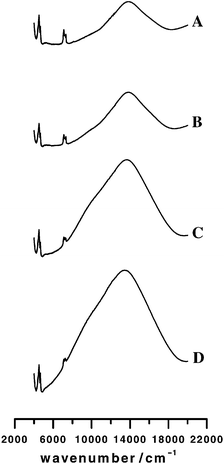 | ||
| Fig. 3 Diffuse reflectance spectra of Cu0.05MOR (A); Cu0.09MOR (B); Cu0.35MOR (C) and Cu0.75MOR (D). | ||
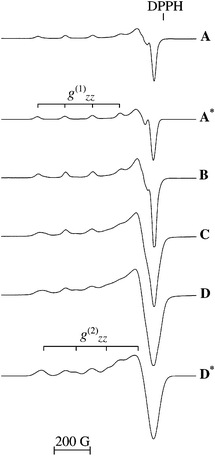 | ||
| Fig. 4 Experimental EPR spectra of Cu0.05MOR (A); Cu0.09MOR (B); Cu0.35MOR (C) and Cu0.75MOR (D). Simulated ESR spectra of Cu0.05MOR (A*) and Cu0.75MOR (D*). | ||
4.2 Cu(II) in site E
The optimized structures of the cluster models representing site E are shown in Fig. 5. Clusters E1–E4 contain only one aluminium, while in clusters E5–E8 two aluminiums were placed at different positions. The corresponding Cu(II) binding energies and the electronic spectra and g-factors obtained from CASPT2 are presented in Table 3. The results for cluster E1.OH will be discussed in section 4.5, together with the other results for (Cu–OH)+ species in rings with one aluminium.| Clusters with one aluminium | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cluster | E1 | E1.OH | E2 | E3 | E4 | |||||
| Binding energy | −508 | — | −491 | −488 | −453 | |||||
| CASPT2 | X 2A | 0 | X 2A | 0 | X 2A | 0 | X 2A″ | 0 | X 2A | 0 |
| spectrum | b 2A | 11623 |
b 2A | 6832 | b 2A | 9728 | a 2A′ | 6366 | b 2A | 8379 |
| c 2A | 13291 |
c 2A | 9570 | c 2A | 11249 |
b 2A′ | 7375 | c 2A | 9417 | |
| d 2A | 13685 |
d 2A | 11577 |
d 2A | 12134 |
b 2A″ | 9471 | d 2A | 10236 |
|
| e 2A | 17311 |
e 2A | 13929 |
e 2A | 15471 |
c 2A′ | 10944 |
e 2A | 13221 |
|
| g-values | 2.28 | 2.43 | 2.32 | 2.45 | 2.36 | |||||
| 2.06 | 2.14 | 2.08 | 2.11 | 2.07 | ||||||
| 2.06 | 2.03 | 2.06 | 2.09 | 2.11 | ||||||
| Clusters with two aluminiums | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cluster | E5 | E6 | E7 | E8 | Experiment | |||||
| Binding energy | −685 | −662 | −649 | −647 | — | |||||
| CASPT2 | X 2A″ | 0 | X 2A | 0 | X 2A | 0 | X 2A | 0 | ||
| spectrum | a 2A′ | 12593 |
b 2A | 8649 | b 2A | 10593 |
b 2A | 10388 |
8000 | |
| b 2A′ | 14224 |
c 2A | 11649 |
c 2A | 11984 |
c 2A | 11465 |
— | ||
| b 2A″ | 14382 |
d 2A | 12239 |
d 2A | 13043 |
d 2A | 12829 |
18000 |
||
| c 2A′ | 16393 |
e 2A | 15355 |
e 2A | 16793 |
e 2A | 15938 |
|||
| g-values | 2.26 | 2.37 | 2.30 | 2.30 | 2.33 | 2.27 | ||||
| 2.06 | 2.09 | 2.10 | 2.08 | 2.07 | 2.09 | |||||
| 2.06 | 2.05 | 2.04 | 2.06 | 2.07 | 2.09 | |||||
From an inspection of the structures E1–E8 in Fig. 5, two general rules concerning the coordination of Cu(II) in zeolites may be deduced, as well as a third more specific rule concerning the coordination at site E. A first general rule is that the local ring distortions, induced by the presence of Cu(II) in one of the zeolite rings, serve to give the latter ion an oxygen coordination number of four. Secondly, Cu(II) prefers to coordinate to oxygens involved in Al–O–Si rather than in Si–O–Si bonds. This second rule is obviously related to the local negative charge connected to the presence of Al in the ring. Thirdly, and specific for site E, Cu(II) coordination in the oxygen six-ring is preferred over coordination in one of the five-rings.
The calculated structures E1–E8 either obey all three or, in case of conflict, at least two of the above rules. Thus, in all but one of the cluster models four short (between 1.90 and 2.25 Å) Cu–O distances are found (rule 1). The only exception is cluster E3, with only three short Cu–O distances. The reason is the strong preference of Cu(II) for the two oxygen neighbours of aluminium (rule 2) at position T2 in cluster E3, far from and therefore inaccessible to other possible oxygen ligands. A strong Cu–O bond with the two Al surrounding oxygens is also observed in clusters E1 and E2, but not in E4, where Al is positioned in the O–T3–O bridge and Cu(II) would therefore have to move from the six-ring to one of the five-rings. Obviously, the third rule prevails in this case, as it does in all clusters in Fig. 5. Actually, a second, local minimum could also be located for cluster E3, with Cu(II) coordinated to four oxygens in one of the bent five-rings. This alternative structure is, however, 4 kcal mol−1 less stable than the structure shown in Fig. 5.
As can be seen from the binding energies in Table 3, by far the strongest Cu–O bonds are formed in clusters E1 and E5, where either one or two aluminiums are placed at the most central T-positions in the six-rings. Indeed, by coordinating to the O–Al–O bonds Cu(II) can, in these clusters, reach an almost perfect square-planar coordination, in agreement with the QM/MM results for a similar site in ZSM-5.17 In cluster E1, an additional interaction is found with one of the oxygens in the O–Si–O bridge, bent inwards and acting as a weak axial ligand (Cu–O distance=2.65 Å), thus giving the Cu(II) ion a five-coordinated square-pyramidal coordination. The large binding energy of Cu(II) in clusters E1, E5 indicates that the central positions of aluminium in site E are optimal for Cu(II)
coordination. The four strong Cu–O bonds also give rise to a ligand field spectrum at high energy, with d–d transitions between 11600 and 17300 cm−1. These results conform with the general spectroscopic characteristics of Cu(II) complexes found in the literature,42 indicating that in general Cu(II) systems with a square-planar or close to square-planar stereochemistry should show d–d transitions at energies higher than 10000 cm−1.42 The close-to-tetragonal symmetry of the E1 and E5 structures is also reflected by their calculated g-factors in Table 3, displaying axial symmetry (gxx=gyy), with gzz values of 2.28 and 2.26 respectively.
Due to the strong tendency of Cu(II) to bind to an O–Al–O unit, more distorted coordination environments are obtained in clusters where aluminium is placed at less central T-sites. Thus one can see how in cluster E2 the Cu–O2A bond is given up (as compared to E1) and a bond between Cu and O1 is formed instead. To accomplish this coordination, O2A is pushed outwards. Similar structures are obtained for clusters E7, E8, with one Al placed as in cluster E2 and the other at one of the T2 sites. In order to embrace the latter Al, Cu(II) would have to form a bond with O5, the least accessible and clearly least favourite oxygen in the ring. The distorted oxygen environment of clusters E2, E7 and E8 weakens the strength of the ligand field. Indeed, in the calculated ligand field spectra (Table 3) the first three excited states are found at significantly (2000 cm−1) lower energies in these clusters than in clusters E1 and E5. The distortion is also reflected in the calculated g-factors: gxx and gyy differ by up to 0.06, while the weakening of the ligand field (i.e. a reduction of the excitation energies in the expression of the g-tensor; see section 3.2) leads to a stronger deviation from ge of gzz, with a calculated value of 2.30–2.32. Even more distorted structures were calculated for the clusters E3, E6 in Fig. 5 while the absence of an aluminium in the six-ring in cluster E4 gives this cluster a regular four-coordinated structure with, however, significantly weaker Cu–O bonds than, for example, in cluster E1. The weaker oxygen ligand field found in the clusters E3, E4 and E6 goes along with a further red-shift of the predicted ligand field excitation energies and with even larger calculated values of gzz, up to 2.45 (Table 3).
4.3 Cu(II) in site C
Site C in mordenite is, like site E, a six-ring with an additional O–T–O bridge. However, the detailed shapes of both sites are quite different. In particular, the oxygen six-ring in site C has a more twisted shape, thus making the site as a whole more compact. On the other hand, one of the oxygen five-rings in site C, i.e. the one located in the main channel of the zeolite (see Fig. 1) is essentially flat, while in site E both five-rings are bent. These structural differences thoroughly affect the coordination properties of Cu(II), as shown by the structures on the different cluster models representing site C, presented in Fig. 6. The corresponding Cu(II) binding energies and the CASPT2 electronic spectra and g-factors are shown in Table 4.| Cluster | C1 | C1.OH | C2 | C3 | C4 | C5 | A1 | Experiment | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Binding energy | −490 | — | −489 | −481 | −490 | −645 | −687 | — | |||
| CASPT2 | X 2A | 0 | 0 | 0 | 0 | 0 | 0 | X 2B | 0 | ||
| spectrum | a 2A | 11042 |
7704 | 11020 |
7004 | 9521 | 6669 | a 2A | 5184 | 8000 | |
| b 2A | 12080 |
9714 | 12118 |
8324 | 10651 |
8916 | b 2A | 6311 | — | ||
| c 2A | 12637 |
11972 |
12742 |
9320 | 11120 |
10096 |
c 2A | 7002 | 18000 |
||
| d 2A | 16780 |
14056 |
16252 |
11167 |
12719 |
12292 |
b 2B | 8603 | |||
| g-values | 2.28 | 2.38 | 2.26 | 2.31 | 2.29 | 2.45 | 2.44 | 2.33 | 2.27 | ||
| 2.07 | 2.14 | 2.07 | 2.30 | 2.17 | 2.05 | 2.10 | 2.07 | 2.09 | |||
| 2.07 | 2.02 | 2.07 | 2.00 | 2.01 | 2.14 | 2.09 | 2.07 | 2.09 | |||
As for site E, two general driving forces determine the structural distortions occurring in the different cluster models of site C upon coordination of Cu(II): the striving of Cu(II) to obtain a four-coordinate square-planar oxygen coordination on the one hand and the preference of this ion for oxygens bound to aluminiums on the other hand. However, as the calculated structures in Fig. 6 indicate, the distortions give rise to different types of Cu(II) coordination environments. Similar to site E, the most regular four-coordinated and also most stable (see the binding energies in Table 4) Cu(II) coordination is obtained in clusters with an aluminium at one of the central T-positions, linking the oxygen five-rings with the six-ring, as in cluster C1. However, the four oxygens making the Cu–O bonds are all part of the planar
five-ring instead of the six-ring (as in site E). This indicates that Cu(II) finds a stable coordination in a flat more easily than in a twisted oxygen-ring. A similar coordination is obtained in structure C2, with an aluminium in the O–T4–O bridge (cf. structure E4 in Fig. 5). We also note that in both clusters C1 and C2 the Cu(II) coordination is pyramidal rather than square-planar. Indeed the Cu(II) ion is located slightly above the plane of the four oxygens towards the main channel of the zeolite (see Fig. 1 and section 5). Even so, the ligand field spectra and g-factors calculated for both structures (Table 4) are similar to the spectra obtained for the square planar structures E1 and E5, with excitation energies ranging between 11000 and
17000 cm−1 and g-factors reflecting axial symmetry with gzz=2.26–2.28.
In the remaining clusters (C3, C4, C5), where aluminium is located at non-central positions in site C, Cu(II) obtains strongly distorted coordination environments in the six-ring. In cluster C3, only two strong Cu–O bonds can be formed. Counting also the two additional weak bonds Cu–O2A
(2.32 Å) and Cu–O2B
(2.27 Å) gives a four-coordinate structure, which is however strongly distorted from the “ideal” square-planar coordination preferred by Cu(II). Similarly distorted structures are also obtained in clusters C4, with an aluminium shared between the six-ring and the twisted five-ring, and in cluster C5, with two aluminiums placed in different five-rings. The strongly distorted character of clusters C3, C4, and C5 is also apparent from their calculated spectral properties (Table 4): the corresponding ligand field
is considerably weaker than for clusters C1 and C2, with excitation energies ranging from 6500 cm−1 up to only 13000 cm−1. Furthermore, the corresponding g-factors are either rhombic (C4, C5) or even predict a “reversed” axial (C3) EPR signal, with two large (2.30–2.31) and one small (2.00)
g-value.
4.4 Cu(II) in site A
The structures and Cu–O distances, resulting from the geometry optimizations of several cluster models representing site A (A1–A5) are presented in Fig. 7. As there are many possibilities for placing one or two aluminiums, we have displayed only the clusters with two aluminiums. In the absence of aluminium and a coordinated cation, the symmetry of this site is C2h. This symmetry is also retained in the structures reported from XRD for Ca2+ coordinated to site A in mordenite.7 This XRD structure was also at the basis of the interpretation, based on AOM-calculations, of one of the signals in the DRS and EPR spectra of Cu(II) in mordenite as belonging to site A.11 Our calculations however strongly indicate that Cu(II) prefers a more asymmetrical coordination in site A. Indeed, even in the absence of any aluminiums, it was found that the Cu(II) ion is moved away from the center of the ring, giving instead a C2 structure with two short (2.05 Å) and two longer (2.55 Å) Cu–O bonds, while the two remaining Cu–O distances have become as long as 3.35 Å. The four Cu–O bonds are considerably shortened (by more than 0.1 Å) when introducing two aluminiums in the ring, connecting the oxygens under consideration, as in cluster A1 represented in Fig. 7. We should also note that the oxygen eight-ring of site A is strongly twisted, such that the Cu(II) coordination is not flat (as might be suggested by the two-dimensional plots in Fig. 7), but should rather be described as distorted tetrahedral; a similar structure was already predicted in a previous, molecular modeling study43 of the Cu(II) coordination in site A, giving Cu–O distances of 2×2.50 and 2×2.73 Å. The corresponding CASPT2 results for the ligand field excitation energies, reported in Table 4 are considerably lower than the excitation energies predicted from AOM: 7200–10700 cm−1versus 11000–14300 cm−1. The CASPT2 excitation energies conform with the more general finding42 that tetrahedral or close-to-tetrahedral Cu(II) complexes absorb primarily in the red and infrared.
In the other clusters in Fig. 7, two aluminiums are positioned such that no symmetry is retained. One can see how in all clusters Cu(II) tries to find a maximum coordination number by choosing, amongst the six central oxygens (obviously, O1 and O5 are not positioned favourably for coordination), the ones that are bound to the aluminiums. As such, either three strong Cu–O bonds or two strong combined with two weak bonds are formed, but a coordination with four strong Cu–O bonds is never reached. It seems that the twisted oxygen eight-ring of site A is simply too large to provide Cu(II) with a planar four-coordination. The electronic spectra of clusters A2–A5 could not be calculated (CASPT2 calculations on clusters of this size fall outside the limits of the presently available MOLCAS code). However, in analogy with cluster A1, the d–d transitions in
the other cluster models may be anticipated to occur at low energies, probably all of them below 10000 cm−1.
4.5 Charge compensation of Cu(II) by OH−
Let us finally look at the two clusters E1.OH and C1.OH, in which an additional OH− counterion is present to compensate for the local negative charge caused by the presence of only one aluminium in this cluster model. These structures are also displayed in Fig. 5 and 6 respectively. As one can see, Cu(II) is, in both structures, coordinated to only three rather than four oxygens: two lattice oxygens and an OH. From a comparison of the original structures of clusters E1 and C1 (in the same figures) with the corresponding OH-containing structures one can see how the Cu(II) ion moves out of the plane of the six- or five-ring and gives up two of the lattice oxygens in favour of one Cu–OH bond, with an exceptionally short Cu–O distance: 1.78–1.79 Å. Not unexpectedly, the two remaining bonds to the lattice are with the oxygens bound to Al. As such, the bonding of the (Cu–OH)+ group in fact reminds one of the bonding of the Cu(I) ion, which is also sometimes found to coordinate to only two lattice oxygens atoms from an AlO4 tetrahedron.14 The three Cu–O bonds are situated in one plane: the sum of the O–Cu–O angles amounts to exactly 360.0° for cluster E1.OH and 359.7° for cluster C1.OH. Hence the geometry of the Cu(II) coordination sphere may be characterized as trigonal planar.It might be of interest to note that a trigonal Cu(II) coordination environment, while very uncommon in inorganic Cu(II) chemistry,42 is also found in a class of proteins called class I or blue copper proteins. The trigonal plane in these proteins is built from two imidazole ligands (from histidine) and a thiolate group SR− (from cysteine). The lack of a fourth ligand in the plane has been explained44,45 by looking at the character of the orbital which is singly occupied in the electronic ground state of the protein. The same considerations may be used to explain the absence of a fourth oxygen ligand in the present OH-containing clusters (with OH− playing a similar role to the SR− group in the proteins). Fig. 8 shows a plot of the singly occupied orbital of the ground state of the E1.OH cluster as compared to the bare E1 cluster. Both orbitals have mainly Cu 3d character. However, in cluster E1, σ-antibonding interactions are found between the four lobes of the Cu 3d orbital and a 2pσ orbital on the lattice oxygens. This antibonding interaction explains how Cu(II) can maximize its ligand field stabilization energy in a square-planar environment (since the corresponding bonding orbital, with predominant oxygen 2pσ character, is doubly occupied). On the other hand, in cluster E1.OH only two of the σ-antibonding interactions remain, while the other two lobes of the Cu 3d orbitals are now instead involved in a π-antibonding interaction with the two lobes of an oxygen 2pπ orbital of the OH− group. The short Cu–OH distance indicates that the latter interaction is very strong, which also explains how it may compensate for the loss of two σ-interactions.
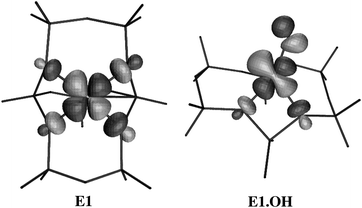 | ||
| Fig. 8 Singly occupied molecular orbital of the ground state of the E1 and E1.OH clusters. | ||
The spectroscopic features of the (Cu–OH)+ species in clusters E1.OH, C1.OH are reported in Tables 3 and 4. As compared to the corresponding results for the bare E1 and C1 clusters (in the same tables), the calculated excitation energies show a pronounced red shift, they are calculated between 6800 and 14000 cm−1. Consequently, the gzz values become much larger, above 2.38. A considerable difference (0.12) is found between gxx and gyy for E1.OH, C1.OH, in contrast to the four-coordinate C1, E1 structures, which were characterized by perfectly axial g-values.
5 Discussion
5.1 Classification of the considered clusters
Although the results of the previous section point to a wide variety of possible coordination modes for Cu(II) to the mordenite framework, it should be clear that in practice only a selected number of these modes will actually occur.46 A critical selection of the most probable coordination sites therefore has to be made. Given the approximate character of the model used for calculating the binding energies (see section 3.1), we believe that these numbers given in Tables 3 and 4 should at most be used as a qualitative tool. An alternative strategy is considered in this work, in which the judgement of the most probable Cu(II) coordination sites is instead based on a confrontation between the calculated spectroscopic properties of the different cluster models (Fig. 5–7) and the information available from the DRS and EPR experiments. In particular, the appearance of only two distinct signals in the EPR spectra is informative, and almost naturally leads to the following classification of the considered clusters.A first class comprises the clusters E1, E5, C1 and C2, i.e. clusters giving rise to a regular, four-coordinated Cu(II) structure with four strong Cu–O bonds of almost equal length: between 1.91 and 2.06 Å. Perfectly axial EPR-signals are predicted for such clusters, with gzz between 2.26 and 2.28 and gxx=gyy=2.06–2.07. The close agreement with the second signal in the EPR spectrum, characterized by gzz=2.27 and gxx=gyy=2.09 strongly suggests that clusters belonging to the first class should be held responsible for this signal. The second class should then collect models connected to the other, first appearing, EPR signal. Approximately
axial g-tensors with gzz values close to 2.33 are calculated only for some of the site E models, i.e. E2, E7, E8 (gzz=2.30–2.32) and possibly also E4 and E6 (gzz=2.36–2.37). As already noted in section 4.2, the less favourable positioning of the aluminiums in these models leads to a generally more distorted Cu–O coordination, but with four Cu–O bonds still formed. As concerns the ligand field transitions, we note that the calculated excitation energies of all models included in both classes considered so far indeed fall within the range of the broad ligand field band in the DRS spectra of Fig. 3
(8000 cm−1–18000 cm−1). This gives further credibility to the proposition that these models may indeed represent the
Cu(II)–zeolite combinations responsible for all experimentally observed spectroscopic features.
For the remaining clusters in Fig. 5–7, EPR signals predicted are not in agreement with the experiments. They are either rhombic (C3, C4) or are characterized by a large value of gzz=2.38–2.45 (E1.OH, C1.OH, E3, C5, A1). The latter goes for clusters E3, C5, A1 along with calculated ligand field excitation energies that fall outside the range of the experimental DRS spectra. We therefore predict that Cu(II)–zeolite combinations represented by this third class of clusters cannot be actually present in Cu-mordenite. We also note that in all these clusters the first coordination sphere of Cu(II) contains either less than four oxygens (clusters E1.OH, C1.OH, E3 and all site A models) or four oxygens in a geometry which is strongly distorted from square-planar (C4, C5).
5.2 Site A
This implies that site A is excluded as a possible site for Cu(II) coordination, the argumentation being that the oxygen eight-ring is too large to supply Cu(II) with the desired oxygen four-coordination. This conclusion is in contradiction with a previous interpretation of de Tavernier,11 holding Cu(II)–site A combinations responsible for the first signal in the EPR-spectrum. Our calculations also seem to contradict the results of the XRD data obtained for Ca-mordenite7 and partially dehydrated Cu-mordenite,8 both pointing to the presence of the respective cations at site A. However, regarding Ca2+, one should bear in mind that this is a much larger ion than Cu(II), and might therefore be more easily accomodated in an oxygen eight-ring. To investigate this further, we performed two additional optimizations on oxygen eight-rings representing site A with either no or two (as in cluster A1) aluminiums, but now containing Ca2+ instead of Cu(II). In the cluster without Al, Ca2+ does not show the tendency of Cu(II) to move towards the side of the ring. C2h symmetry is retained, and Ca2+ is found to be six-coordinated, although the Ca–O distances (2×2.47, 4×2.64 Å) are larger than the Ca–O distance of 2.33 Å found for e.g. Ca2+ surrounded by six H2O.47 As compared to the XRD data for Ca-mordenite7 the calculated distances are slightly shorter: 2.47 versus 2.54 Å
(XRD) and 2.64 versus 2.85 Å
(XRD). In the cluster with two Al (A1.Ca in Fig. 7), Ca2+ also prefers to bind to the O–Al–O sequences and therefore moves away from the center of the ring. However, it is clear that much less severe distortions of the oxygen eight-ring are required to comfortably host Ca2+ than for Cu(II).
In partially dehydrated Cu(II) MOR, Cu(II) ions are also found in site A.8 However, they are coordinated to two water ligands. The structures resulting from geometry optimizations of two clusters of site A with two Al and with either one (Al·H2O) or two (Al·(H2O)2) additional water ligands are also included in Fig. 8. They show how the Cu(II) ion can easily complement its first coordination sphere with the extra H2O to obtain a total number of four or five ligands. Our argument for rejecting the presence of Cu(II) in site A therefore does not hold in the presence of H2O. This does, however, imply the migration of Cu(II) ions away from site A during the dehydration proces.
In the XRD structure of partially dehydrated Cu(II) MOR, a very small portion of the Cu(II) ions were also located in site D. However, since the oxygens in this eight-ring are even further apart than in the twisted eight-ring of site A (the shortest distance between opposite oxygens being 6.28 Å in site D as compared to 5.14 Å in site A) it is highly unlikely that Cu(II) would be able to obtain an oxygen four-coordination in site D, at least not without extra H2O ligands. Finally, it should be noted that the cation exchange level in all XRD studies is much higher than the Cu(II) contents considered in this work.
5.3 Interpretation of the two EPR signals
Even if the above classification can explain the presence of two signals in the EPR spectrum of Cu(II) mordenite, additional arguments are needed to explain the order in which the two signals appear. Indeed, the fact that the second EPR signal is connected to clusters with a regular square-planar or square-pyramidal Cu(II) coordination sphere, while the first signal corresponds to a more distorted coordination implies that Cu(II) actually prefers the less ideal coordination structure, giving rise to a weaker ligand field. To resolve this seeming contradiction the following two extra factors should be taken into account: (i) electrostatic factors dictate that Cu(II) will preferentially coordinate to oxygen rings containing two instead of only one Al, and (ii) two Al cannot be contained in an oxygen five-ring. The latter rule was introduced by Takaishi et al.25,26 based on 29Si NMR results and considering the connectivity relations between T-sites in mordenite. According to this rule two of the calculated Al distributions in site E, i.e. clusters E5, E6 in Fig. 5, cannot occur in the actual zeolite. From the binding energies in Table 3 we note that these two clusters, in particular E5, give a considerably stronger bond with Cu(II) than the other two clusters with two Al, E7 and E8. Also note that for site C, clusters violating Takaishi's rule were never even considered. For example, a cluster giving a regular square-pyramidal coordination with four strong bonds might also here have been designed by starting from cluster C1, and placing a second aluminium in the planar five-ring, opposite to the Al already present. Hence Takaishi's rule in fact a priori excludes those aluminium distributions that might have provided Cu(II) with a regular square-planar or square-pyramidal coordination in a ring containing two aluminiums, thereby giving rise to a gzz value of around 2.27. The preferential coordination of Cu(II) to oxygen rings containing two rather than only one aluminium is dictated by electrostatic factors but occurs at the expense of having to settle for a less favourite electronic bonding situation, i.e. a smaller ligand field stabilization energy, and a concomitant higher value of gzz.Our interpretation of the two signals in the EPR spectrum of Cu(II) mordenite partly agrees with a previous interpretation given by Wichterlová
et al., which also related the EPR signals with gzz=2.33 and 2.27 to Cu(II) in the vicinity of two and only one aluminium respectively.12 However, the connection made by Wichterlová
et al. between the value of gzz, 2.33 or 2.27, on the one hand and the structure of the Cu(II) oxygen ligand environment, i.e. square-pyramidal or square-planar, on the other hand, is not confirmed by our calculations. Indeed, a value of gzz=2.26–2.28 is calculated both for a square-planar (such as E1) and a square-pyramidal coordination (as in C1, C2). A more quantitative measure
of the “pyramidality” of the Cu–oxygen coordination at a given site may be obtained by summing the four O–Cu–O angles within the first coordination sphere. Thus we obtain a sum of 351.4° for cluster E1, indicating that Cu(II) is indeed situated almost in the plane of the four oxygens, while considerably smaller values, 334.0 and 331.5, are obtained for clusters C1, C2, indicating a more pyramidal structure. However, values in between those two extremes are found for the clusters giving rise to larger gzz values, e.g. 346.0 for cluster E7 and 344.7 for cluster E8. This confirms the absence of a relation between the EPR g-factors and the pyramidality of the Cu(II)–oxygen environment. As already seen before, the larger gzz value should rather be connected to a more irregular four-fold coordination, characterized for example by one exceptionally
large O–Cu–O angle, i.e. 108.1° in cluster E7 and 111.7° in cluster E8.
5.4 Charge compensation of Cu(II)
We have also concluded that our Cu(II)-mordenite samples do not contain any significant amount of (Cu–OH)+ species, as the gzz values calculated for these coordination modes were not in agreement with experiment. It should, however, be stressed that this conclusion might only be valid for the range of copper loadings considered in this work, and that (Cu–OH)+ species could still appear at higher loadings or in the presence of water. More symmetrical four-coordinate structures can indeed result when adding one water molecule to clusters E1.OH and C1.OH, as was demonstrated by the results of some additional geometry optimizations.The maximum considered Cu/Al ratio of 0.11 is probably low enough to allow for compensation of the excess charge connected to a Cu(II) in a ring with only one Al by another nearby Al. That this might indeed be the case is in fact indicated by the 29Si NMR data (section 4.1), showing that each unit cell should contain about 2.5 Al–O–Si–O–Al links, i.e. more than three times the maximum loading considered in this work, of 0.75 Cu(II) per unit cell. This strongly suggests that in cases where Cu(II) is bound to a ring containing one Al, the latter is in fact connected by an O–Si–O bridge to another Al outside the ring. Furthermore, Al–(Si)n–Al sequences with n>1 might also balance the charge of divalent cations.
6 Concluding remarks
In the present contribution we have shown how information obtained from DRS and EPR spectra of Cu(II) in mordenite may be successfully combined with quantum chemical cluster model calculations to obtain information about the siting and coordination of Cu(II) ions in mordenite. In particular, the confrontation between experimental and calculated g-factors has proven to be useful, giving valuable insight into the factors that govern the bonding of the ion at different sites. The EPR signal with gzz=2.27 has been attributed to regular four-coordinated square-planar or square-pyramidal lattice oxygen environments, while the larger gzz value of 2.33 was shown to be connected to a more distorted oxygen environment, giving rise to a weaker ligand field. It has been shown how electrostatic effects are important, directing the first Cu(II) ions
to sites containing two Al. Takaishi's rule has been invoked to explain the absence of the EPR-signal with gzz=2.27 in connection with such rings.
Our calculations have led to the general observation that, next to electrostatic effects, the striving of a Cu(II) ion for a square-planar coordination plays a crucial role in determining the possible coordination sites in the zeolites. We have shown how coordination in planar six-rings (site E) or five-rings (site C) is strongly preferred, whereas coordination in twisted or too large rings (site A) is avoided. The following sites have come out of the analysis as most probable coordination sites at low Cu(II) loadings: E7 and E8, both containing two Al, and giving rise to the EPR signal with gzz=2.33, and E1, C1 and C2, containing one Al and responsible for the signal with gzz=2.27. Cluster E2 (one Al) is also not excluded as a secondary source for the gzz=2.33
signal. It should be noted that both the planar six-ring of site E and the planar five-ring of site C are located in the main channel of the zeolite. This is in accord with a previous EPR study,9 indicating that in mordenite all Cu(II) ions should indeed be located at sites in the main channel, since they can interact with large molecules such as benzene and o-xylene. Our calculations have also indicated that even in rings containing only one aluminium Cu(II) is coordinated as a single ion, not as (Cu–OH)+.
Finally, the question might be raised whether the geometrical distortions of the actual framework rings in the zeolite caused by the Cu(II) ions (and reflected, for example, by the asymmetric vibrations of the T–O bonds48) can really be accurately predicted with the limited cluster models used in this work. We believe that this is indeed the case, at least for the oxygen six- and five-rings at sites C and E. Previous calculations on similar cluster models for Cu(II) coordinated at oxygen six-rings in the zeolites A, Y and ZK4 have proven that also in this case the experimental DRS spectra (showing a much higher resolution than in mordenite) as well as the EPR g-tensors could be accurately reproduced.18 It was also shown in this work that the calculated values for these spectroscopic features are critically dependent on the geometry of the cluster models. The close correspondence between the experimental spectroscopic data for the zeolites and the calculated data for the model clusters therefore offers a good indication that the structure of the model clusters is indeed close to the actual geometry of the Cu(II) surrounding in the zeolites. However, as concerns the oxygen eight-rings at site A in mordenite our conclusion should be more restrictive. It cannot be excluded that the size of the cluster model presently used is too small to allow for the full geometrical distortions necessary for Cu(II) to obtain a four-coordinated oxygen environment with four short Cu–O bonds, and that the use of larger models or embedded clusters might indeed lead to further relaxation of the structure. This will be the subject of further investigation by our group.
Acknowledgements
Prof. P. J. Grobet is kindly acknowledged for the performance of the 29Si NMR measurement. This investigation has been supported by grants from the Flemish Science Foundation (FWO) and from the Concerted Research Action of the Flemish Government (GOA). M. G. thanks the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT) for a research grant.References
- W. J. Mortier and R. A. Schoonheydt, Prog. Solid State Chem., 1985, 16 CrossRef CAS.
- W. J. Mortier, Compilation of Extra-Framework Sites in Zeolites, Butterworth, London, 1982. Search PubMed.
- W. M. Meier, D. H. Olson and C. Baerlocher, Atlas of Zeolite Structure Types, Elsevier, Amsterdam, 4th edn., 1996. Search PubMed.
- M. Iwamoto, H. Furukawa, Y. Mine, F. Uemura, S. Mikuriya and S. Kagawa, J. Chem. Soc., Chem. Commun., 1986, 1273 RSC.
- M. Shelef, Chem. Rev., 1995, 95, 209 CrossRef CAS.
- E.-Y. Choi, I.-S. Nam and Y. G. Kim, J. Catal., 1996, 161, 597 CrossRef CAS.
- W. J. Mortier, J. J. Pluth and V. J. Smith, Mater. Res. Bull., 1975, 10, 1037 CrossRef CAS.
- M. P. Attfield, S. J. Weigel and A. K. Cheetham, J. Catal., 1997, 170, 227 CrossRef CAS.
- A. V. Kucherov, A. A. Slinkin, D. A. Kondrat'ev, T. N. Bondarenko, A. M. Rubinstein and K. M. Minachev, Zeolites, 1985, 5, 320 CrossRef CAS.
- C. E. Sass and L. Kevan, J. Phys. Chem., 1989, 93, 4669 CrossRef CAS.
- S. de Tavernier and R. A. Schoonheydt, Zeolites, 1991, 11, 155 CrossRef CAS.
- J. Dĕde
![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) ek, Z. Sobalík, Z. Tvarů
ek, Z. Sobalík, Z. Tvarů![[z with combining breve]](https://www.rsc.org/images/entities/char_007a_0306.gif) ková, D. Kaucký and B. Wichterlová, J. Phys. Chem., 1995, 99, 16327 CAS.
ková, D. Kaucký and B. Wichterlová, J. Phys. Chem., 1995, 99, 16327 CAS. - B. Wichterlová, Z. Sobalík and A. Vondrová, Catal. Today, 1996, 29, 149 CrossRef CAS.
- D. Nachtigallová, P. Nachtigall, M. Sierka and J. Sauer, Phys. Chem. Chem. Phys., 1999, 1, 2019 RSC.
- M. Sierka and J. Sauer, Faraday Discuss., 1997, 106, 41 RSC.
- J. Sauer, Chem. Rev., 1989, 89, 199 CrossRef CAS.
- D. Nachtigallová, P. Nachtigall and J. Sauer, Phys. Chem. Chem. Phys., 2001, 3, 1552 RSC.
- K. Pierloot, A. Delabie, M. H. Groothaert and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 2001, 3, 2174 RSC.
- K. Pierloot, A. Delabie, C. Ribbing, A. A. Verberckmoes and R. A. Schoonheydt, J. Phys. Chem. B, 1998, 102, 10789 CrossRef CAS.
- K. Pierloot, A. Delabie, A. A. Verberckmoes and R. A. Schoonheydt, in Density Functional Theory, a Bridge between Chemistry and Physics, ed. P. Geerlings, F. D. Proft and W. Langenaeker, VUB University Press, Brussels, 1999, pp. 169–188. Search PubMed.
- A. Delabie, K. Pierloot, M. H. Groothaert, B. M. Weckhuysen and R. A. Schoonheydt, Microporous Mesoporous Mater., 2000, 37, 209 CrossRef CAS.
- Y. Kuroda, A. Kotani, H. Maeda, H. Moriwaki, T. Morimato and M. Nagao, J. Chem. Soc., Faraday Trans., 1992, 88, 1583 RSC.
- Illinois EPR Research Center, Urbana-Champaign, USA. Simpow program..
- W. Loewenstein, Am. Miner., 1942, 39, 92 Search PubMed.
- T. Takaishi, M. Kato and K. Itabashi, Zeolites, 1995, 15, 21 CrossRef CAS.
- T. Takaishi, M. Kato and K. Itabashi, J. Phys. Chem., 1994, 98, 5742 CrossRef.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- B. L. Trout, A. K. Chakraborty and A. T. Bell, J. Phys. Chem., 1996, 100, 4173 CrossRef CAS.
- P. J. Carl and S. C. Larsen, J. Phys. Chem. B, 2000, 104, 6568 CrossRef CAS.
- M. L. Jacono, G. Fierro, R. Dragone, X. Feng, J. d’Itri and W. K. Hall, J. Phys. Chem. B, 1997, 101, 1979 CrossRef.
- Previous work has demonstrated that the interaction between the Cu-site and one ligand can succesfully be described by our cluster models, see ref. 21..
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218 CrossRef CAS.
- K. Andersson, M. R. A. Blomberg, M. P. Fülscher, G. Karlström, R. Lindh, P.-Å. Malmqvist, P. Neogrády, J. Olsen, B. O. Roos, A. J. Sadlej, M. Schütz, L. Seijo, L. Serrano-Andrés, P. E. M. Siegbahn and P.-O. Widmark, MOLCAS Version 4.0. Department of Theoretical Chemistry, University of Lund, Sweden, 1997. Search PubMed.
- K. Pierloot, B. Dumez, P.-O. Widmark and B. O. Roos, Theor. Chim. Acta, 1995, 90, 87 CrossRef CAS.
- B. O. Roos and K. Andersson, Chem. Phys. Lett., 1995, 245, 215 CrossRef CAS.
- C. Ribbing and C. Daniel, J. Chem. Phys., 1994, 100, 6591 CrossRef CAS.
- N. M. Atherton, Electron Spin Resonance, Theory and Applications, Ellis Horwood Limited, Chichester, 1973. Search PubMed.
- Since the non-equivalency of the 4 T-sites in a mordenite causes only small differences in the 29Si chemical shifts, there is no contribution of Si(0Al) to the Si(1Al) peak or vice versa, see ref. 40..
- G. Engelhardt and D. Michel, High-resolution Solid-state NMR of Silicates and Zeolites, Wiley, Chichester, 1986. Search PubMed.
- R. A. Schoonheydt, Catal. Rev.-Sci. Eng., 1993, 35, 129 Search PubMed.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1984. Search PubMed.
- V. Guliants, J. Mullhaupt, J. Newsam, A. Gorman and C. Freeman, Catal. Today, 1999, 50, 661 CrossRef CAS.
- K. Pierloot, J. O. A. De Kerpel, U. Ryde, M. H. M. Olsson and B. O. Roos, J. Am. Chem. Soc., 1998, 120, 13156 CrossRef CAS.
- M. H. M. Olsson, U. Ryde, B. O. Roos and K. Pierloot, J. Biol. Inorg. Chem., 1998, 3, 109 CrossRef CAS.
- This is the more true since the samples used in this work have quite low copper loadings, the maximum loading of 0.75 Cu per unit cell (2(Cu/Al)=21%) remaining well below the estimated Cu(II) content of 3.5 Cu per unit cell of a totally exchanged CuMOR (2(Cu/Al)=100% and with Si/Al=5.7, as obtained from NMR)..
- O. Carugo, K. Djinovic and M. Rizzi, J. Chem. Soc., Dalton Trans., 1993, 2127 RSC.
- Z. Sobalík, Z. Tvarůková and B. Wichterlová, J. Phys. Chem. B, 1998, 102, 1077 CrossRef CAS.
| This journal is © the Owner Societies 2002 |