Determination of the basicity of alkali-exchanged molecular sieves
Georgi N.
Vayssilov
*ab and
Notker
Rösch
*a
aInstitut für Physikalische und Theoretische Chemie, Technische Universität München, 85747 Garching, Germany. E-mail: roesch@ch.tum.de
bFaculty of Chemistry, University of Sofia, 1126 Sofia, Bulgaria. E-mail: gnv@chem.uni-sofia.bg
First published on 17th December 2001
Abstract
A procedure is proposed to quantify the basicity of zeolite oxygen centers by estimating the proton affinity (PA) of these centers, employing measured IR frequency shifts of the stretching and deformation modes of the OH group of adsorbed methanol. The PA scale is established with the help of density functional theory (DFT) calculated PA values of selected zeolites calibrated to the experimental gas phase PA of small oxygen-containing molecules. The reliability of the method is increased by combining two experimental parameters.
Introduction
The basicity of oxygen centers in zeolites plays an important role in their catalytic and adsorption properties.1–3 For instance, alkali-exchanged forms of zeolites are utilized as basic catalysts in the side-chain alkylation of toluene as well as in condensation and cyclo-condensation reactions.1,4,5 In addition, the framework basicity is important in processes that require catalytic acid–base pairs, e.g. alcohol dehydrogenation or selective sorption and separation.1,6,7One of the criteria for basicity is the 1s core level energy, Eb(O 1s), of the oxygen centers in zeolites, as measured by XPS. Indeed, we have recently reported a linear correlation between calculated values of the proton affinity (PA) of an oxygen center in a zeolite and the corresponding O 1s core level shift ΔEb(O 1s) relative to a reference.8 The accuracy of XPS, however, does not allow one to discriminate different oxygen centers in a zeolite; rather, the measured O 1s binding energy (shift) is an average value over all types of oxygen centers of the material. Thus, Eb(O 1s) is useful as a basicity measure only for materials with uniform oxygen centers, e.g. zeolites with Si only as T-atoms or with an Al/Si ratio close to one. Other basicity criteria exploit spectral properties of adsorbed probe molecules, e.g. IR spectra of adsorbed chloroform,9 pyrrole,10 methanol,11–14 or 13C NMR of surface methoxy groups.15,16 However, it is not always clear whether variations of a spectral property originate from the interaction of the probe with a basic site or with charge-compensating metal cations. In addition, there is no unified quantitative measure for basicity; rather, each spectral method defines its own scale for the relative basicity of the investigated materials.1
A direct absolute measure of zeolite basicity is the PA of an oxygen center, i.e. the energy gained after attachment of a proton to the basic site under study. The PA value is related to both Eb(O 1s)8 and, via the hydrogen bond formed between the adsorbate and the framework oxygen centers, to IR frequency shifts. In the following, we will focus on the latter relationship with methanol as a probe molecule. In recent density functional theory (DFT) calculations17 we demonstrated that calculated O–H vibrational frequency shifts of methanol adsorbed on alkali-exchanged zeolites are particularly sensitive to the basicity of zeolite oxygen centers and that they correlate in linear fashion with their calculated PA. To render this correlation directly applicable to experiment without the detour to calculations, we calibrated the calculated PA using experimental gas phase PA values18 of a series of small oxygen-containing organic molecules and water. Based on these modified PA values, we derived equations providing an estimate for the measured PA of the basic zeolite oxygen centers using the frequency shift of stretching and deformation OH bands of adsorbed methanol.
Method
Density functional calculations were performed with the program PARAGAUSS19,20 using a gradient-corrected exchange-correlation functional.21,22 The Kohn–Sham orbitals were represented by Gaussian-type basis sets, contracted in generalized form: (6s1p)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) →[3s1p] for H, (9s5p1d)→[5s4p1d] for O and C, (12s9p1d)→[6s4p1d] for Al and Si, (12s8p1d)→
[6s5p1d] for Na, and (15s11p1d)→[6s5p1d] for K.23 The alkali cations were located at a six-ring faujasite structure with one or two Al centers. The geometry of the zeolite clusters was optimized using analytical energy gradients, but T atoms were constrained to experimentally determined positions. The geometry of the
reference organic molecules was optimized without constraints. The PA values of molecules were determined as total energy differences between the neutral species and the molecule protonated at an oxygen center, using optimized structures in each case. The PA values of zeolite oxygen centers were obtained in the same fashion.
→[3s1p] for H, (9s5p1d)→[5s4p1d] for O and C, (12s9p1d)→[6s4p1d] for Al and Si, (12s8p1d)→
[6s5p1d] for Na, and (15s11p1d)→[6s5p1d] for K.23 The alkali cations were located at a six-ring faujasite structure with one or two Al centers. The geometry of the zeolite clusters was optimized using analytical energy gradients, but T atoms were constrained to experimentally determined positions. The geometry of the
reference organic molecules was optimized without constraints. The PA values of molecules were determined as total energy differences between the neutral species and the molecule protonated at an oxygen center, using optimized structures in each case. The PA values of zeolite oxygen centers were obtained in the same fashion.
Results and discussion
The calculated and experimental PA values of free organic molecules and water are shown in Table 1. As a general trend, the calculations at that level overestimate the PA with respect to the experiment on average by 27 kJ mol−1. According to this trend, the calculated PA values of the zeolite oxygen centers should be by adjusted by −27 kJ mol−1 to obtain more accurate estimates of measured PA values.mol−1) of reference molecules
Based on the linear relationship shown in Fig. 1 (including corrected PA values), we here propose to estimate experimental PA values of zeolite oxygen centers from measured IR spectra of methanol adsorbed on alkali-exchanged zeolites, using the frequency shifts of the methanol O–H stretching and deformation bands, Δν(OH) and Δδ(OH), respectively:
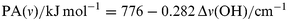 | (a) |
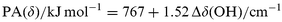 | (b) |
=772±5 kJ mol−1.
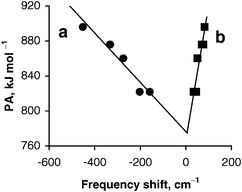 | ||
| Fig. 1 Correlation between DFT calculated PA values of oxygen centers of zeolites NaY, NaX and KX (corrected by −27 kJ mol−1) and the corresponding experimental frequency shifts of the stretching (Δν(OH), a) and deformation (Δδ(OH), b) bands of adsorbed methanol. The correlation coefficients of the straight lines are 0.94 (a) and 0.89 (b). | ||
The correlations in Fig. 1 are derived from model cluster calculations on O,H-bound species17 where the methanol probe coordinates to an alkali cation and, in addition, forms a hydrogen bond with a nearby oxygen center. Such a structure reflects the situation of methanol adsorption complexes for which the experimental frequencies have been measured.24 Although the interaction of methanol with the cation affects the frequency shifts of the stretching and deformation vibrations, the hydrogen bond of methanol with the zeolite oxygen centers exerts by far the largest effect on these frequencies.17 This can be seen by comparing the frequency shifts calculated for adsorption complexes of O,H-bound probe molecules with those of structures where methanol binds only via its oxygen center to the alkali cation. In the latter case, the frequency shifts are 3–10 times smaller.17 The correlation of the experimentally measured frequency shifts with the calculated PA of zeolite oxygen centers (Fig. 1) also shows that the effect of the hydrogen bond dominates the OH frequency shifts.
The PA values determined from eqn. (a) and (b) account for the basicity of oxygen centers in the vicinity of a metal cation. This is useful because those centers are considered responsible for the catalytic and sorptive properties of alkali-exchanged zeolites.1,24–26
The proposed approach permits the assignment of PA values to oxygen centers of different basicity in a zeolite sample8 if the IR spectrum exhibits distinguishable frequency shifts Δν(OH) and Δδ(OH). Since the formation of hydrogen bonds between two adsorbed methanol molecules also contributes to the splitting of the OH bands,24 the frequency shifts Δν(OH) and Δδ(OH) should be measured at very low methanol coverage.
As an example, we illustrate the proposed procedure with measured IR spectra of methanol adsorbed on various zeolite samples.24 First, we focus on the PA of oxygen centers in faujasites, NaY, NaX, KX, and RbX (Table 2). The values of the former three samples were used in the derivation of eqn. (a) and (b). In the IR spectra of methanol adsorbed on RbX zeolite, there are two stretching and two deformation bands for hydrogen-bound OH groups.24 Thus, one expects two types of oxygen centers to be involved in hydrogen bonding. Their frequency-derived average PA values are 861 and 909 kJ mol−1, respectively (Table 2). From the differences ΔPA of the two values (a) and (b) for each type of basic center an inherent uncertainty of at most 13 kJ mol−1 is derived.
mol−1), of oxygen centers in various alkali-exchanged zeolites derived from measured OH vibrational frequency shifts via eqn. (a) and (b). Also shown are the differences ΔPA of the values (a) and (b) as well as the underlying experimental frequency shifts Δν and Δδ
(in cm−1)
| Sample | Si/Ala | Δνa | Δδa | PAb | ΔPAc | PA(DFT)d |
|---|---|---|---|---|---|---|
|
a Si/A1 ratio and IR frequency shifts according to ref. 24.
b Average of PA(ν) and PA(δ) calculated from eqn. (a) and (b).
c ΔPA=PA(ν)−PA(δ).
d From DFT calculations, ref. 8, corrected by −27kJmol−1.
e
Eqn. (a) and (b) established using data of these samples.
|
||||||
| NaZSM-5 | 34.6 | −75 | 17 | 795 | 5 | |
| Na-MOR20 | 10.1 | −82 | 15 | 795 | 10 | |
| −212 | 65 | 851 | −30 | |||
| Na-MOR10 | 4.9 | −103 | 25 | 805 | 1 | |
| −209 | 67 | 852 | −33 | |||
| −339 | 67 | 870 | 3 | |||
| NaYe | 2.8 | −158 | 35 | 821 | 1 | 822 |
| −332 | 77 | 877 | −14 | 876 | ||
| NaXe | 1.3 | −202 | 45 | 834 | −2 | 822 |
| −333 | 70 | 872 | −3 | 876 | ||
| KXe | 1.3 | −274 | 51 | 849 | 9 | 860 |
| −452 | 82 | 898 | 12 | 896 | ||
| RbX | 1.3 | −278 | 66 | 861 | −13 | |
| −472 | 93 | 909 | 1 | |||
Almost all oxygen centers of X zeolites participate in Al–O–Si bridges since the Si/Al ratio is close to one. Therefore, contributions of oxygen centers in Si–O–Si bridges (not included in the frequency-derived PA values) to measured O1s core levels are negligible and we may check the IR-based PA values by XPS measurements. Accordingly, the O1s core levels of RbX are on average 0.3–0.5 eV less stable than the average O1s core levels of NaX.1,9 The PA value has been calculated to decrease by 8 kJ mol−1 when the calculated core level binding energy Eb(O1s) of the zeolite oxygen center increases by 0.1 eV (stabilization of the core levels).8 Thus, from the XPS results one expects the PA values of RbX to be about 24–40 kJ mol−1 higher than those of NaX. This fits the basicity differences, 27–37 kJ mol−1, derived from the experimental IR spectra of adsorbed methanol very well (Table 2).
For most of the bands of methanol adsorbed on Na-exchanged ZSM-5 and the mordenite samples Na-MOR10 and Na-MOR20 (the numbers denote the SiO2/Al2O3 ratio), eqn. (a) and (b) yield very similar PA values, the differences ΔPA being at most 10 kJ mol−1
(Table 2). This finding corroborates the stability of the proposed PA criterion. However, the PA of the zeolite oxygen centers responsible for the band with Δν(OH)=−209 cm−1 on both mordenite samples differs by about 30 kJ mol−1 when stretching or deformation O–H frequency shifts are used (Table 2). This has been identified17 as an indication of steric restrictions acting on the methanol
methyl group which affect the deformation O–H mode. Thus, the combined application of eqn. (a) and (b) is limited to zeolite systems where no serious steric constraints occur at the adsorption site; otherwise, the linear correlation of the frequency shifts Δδ(OH) can be perturbed.
The obtained PA scale complies well with known catalytic properties of alkali-exchanged zeolites. For example, the selectivity to side-chain alkylation of toluene, catalyzed by basic centers, is 3% for NaX zeolite and increases to 46 and 65% for KX and RbX, respectively.26 From Table 2, one notes PA estimates of the oxygen centers in KX and RbX zeolites that are about 20(±5) and 32(±5) kJ mol−1 higher than those of the corresponding basic centers of NaX. The reaction rate constant of the base-catalyzed Knoevenagel condensation on similar zeolite samples5 was also observed to increase in the order predicted by the PA estimates.
Alternatives
The use of other probe molecules is conceivable as is a transfer of the suggested strategy to other materials with basic centers. For example, trifluoromethane could be a convenient probe because neither of the IR bands ν(CH) and δ(CH) is covered by the absorbance of the zeolite framework;9 of course, suitable theoretical data on the adsorption of this molecule on alkali zeolites have to be generated first. Further extension of the proposed approach with other probe molecules and spectral features should allow the construction of a general scale for zeolite basicity.Kaliaguine and co-workers10 suggested another combined approach to the basicity of zeolites, based on the N–H frequency and the N 1s core level shift of adsorbed pyrrole molecules. However, it is not clear whether the good correlation between these quantities is affected by the formation of a hydrogen bond with basic sites or is mainly influenced by the electrostatic field of cations in the zeolite. Unfortunately, a recent computational study27 of pyrrole adsorption on alkali zeolites did not consider this problem. The N–H deformation band of adsorbed pyrrole is not suitable as a second measure because it overlaps (at about 1100 cm−1) with a strong absorption of the zeolite framework. After careful examination of the factors influencing 13C chemical shifts of magic angle spinning (MAS) NMR spectra of adsorbed methoxy groups on zeolites, such spectroscopic data could also be employed for constructing a basicity scale.
Acknowledgements
We thank Professor J. A. Lercher and Professor M. Hunger for helpful discussions. G.N.V. gratefully acknowledges a fellowship of the Alexander von Humboldt Foundation and an equipment donation. This work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.References
- D. Barthomeuf, Catal. Rev., 1996, 38, 521 Search PubMed.
- H. Hattori, Chem. Rev., 1995, 95, 537 CrossRef CAS.
- K. Tanabe and W. F. Hölderich, Appl. Catal. A, 1999, 181, 399 CrossRef CAS.
- P. E. Hathaway and M. E. Davis, J. Catal., 1989, 119, 497 CrossRef CAS.
- A. Corma, V. Fornes, R. M. Martin-Aranda, H. Garcia and J. Primo, Appl. Catal., 1990, 59, 237 CrossRef CAS.
- C. Bezoukhanova and Yu. Kalvachev, Catal. Rev., 1994, 36, 125 Search PubMed.
- D. R. Corbin and B. A. Mahler, World Pat. W.O. 94/02440, 1994..
- G. N. Vayssilov and N. Rösch, J. Catal., 1999, 186, 423 CrossRef CAS.
- E. Bosch, S. Huber, J. Weitkamp and H. Knözinger, Phys. Chem. Chem. Phys., 1999, 1, 579 RSC.
- M. Huang, A. Adnot and S. Kaliaguine, J. Catal., 1992, 137, 322 CrossRef CAS.
- A. E. Palomares, G. Eder-Mirth and J. A. Lercher, J. Catal., 1997, 168, 442 CrossRef CAS.
- G. Busca, P. F. Rossi, V. Lorenzelli, M. Benaissa, J. Travert and J. C. Lavalley, J. Phys. Chem., 1985, 89, 5433 CrossRef CAS.
- M. Ziolek, J. Czyzniewska, J. Lamotte and J. C. Lavalley, Catal. Lett., 1996, 37, 223 Search PubMed.
- B. Hunger, S. Matysik, M. Heuchel and W. D. Einicke, Langmuir, 1997, 13, 6249 CrossRef CAS.
- V. Bosáček, J. Phys. Chem., 1993, 97, 10732 CAS.
- U. Schenk, M. Hunger and J. Weitkamp, Magn. Reson. Chem., 1999, 37, S75 CrossRef CAS.
- G. N. Vayssilov, J. A. Lercher and N. Rösch, J. Phys. Chem. B, 2000, 104, 8614 CrossRef CAS.
- B. E. Mills, R. L. Martin and D. A. Shirley, J. Am. Chem. Soc., 1976, 98, 2380 CrossRef CAS.
- T. Belling, T. Grauschopf, S. Krüger, M. Mayer, F. Nörtemann, M. Staufer, C. Zenger and N. Rösch, in High Performance Scientific and Engineering Computing, Lecture Notes in Computational Science and Engineering, ed. H.-J. In Bungartz, F. Durst and C. Zenger, Springer, Heidelberg, 1999, vol. 8, p. 439. Search PubMed.
- PARAGAUSS version 2.1, T. Belling, T. Grauschopf, S. Krüger, F. Nörtemann, M. Staufer, M. Mayer, V. A. Nasluzov, U. Birkenheuer, A. Hu, A. V. Matveev and N. Rösch, Technische Universität München, München, 2000..
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- (a) J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef; (b) J. P. Perdew, Phys. Rev. B, 1986, 34, 7406 CrossRef.
- A. M. Ferrari, K. M. Neyman and N. Rösch, J. Phys. Chem. B, 1997, 101, 9292 CrossRef CAS.
- M. Rep, A. E. Palomares, G. Eder-Mirth, J. G. van Ommen, N. Rösch and J. A. Lercher, J. Phys. Chem. B, 2000, 104, 8624 CrossRef CAS.
- J. F. Goellner, B. C. Gates, G. N. Vayssilov and N. Rösch, J. Am. Chem. Soc., 2000, 122, 8056 CrossRef CAS.
- A. E. Palomares, G. Eder-Mirth, M. Rep and J. A. Lercher, J. Catal., 1998, 180, 56 CrossRef CAS.
- H. Föster, H. Fuess, E. Geidel, B. Hunger, H. Jobic, C. Kirschhock, O. Klepel and K. Krause, Phys. Chem. Chem. Phys., 1999, 1, 593 RSC.
| This journal is © the Owner Societies 2002 |