Effects of thermal pretreatment on the redox behaviour of Ce0.5Zr0.5O2: isotopic and spectroscopic studies
P.
Fornasiero
a,
T.
Montini
a,
M.
Graziani
a,
J.
Kašpar
*a,
A. B.
Hungría
b,
A.
Martínez-Arias
b and
J. C.
Conesa
*b
aDipartimento di Scienze Chimiche, Università di Trieste, via Giorgieri 1, 34127 Trieste, Italy. E-mail: kaspar@univ.trieste.it
bInstituto de Catálisis y Petroleoquímica, CSIC. Campus de Cantoblanco, 28049 Madrid, Spain. E-mail: jcconesa@icp.csic.es
First published on 17th December 2001
Abstract
The temperature programmed reduction (TPR) of a single phase Ce0.5Zr0.5O2 solid solution is investigated by using 1H2/Ar and 2H2/Ar mixtures as reducing agents. The effects of mild (700 K) or severe (1273 K) pre-oxidation (MO, SO) on the TPR profile are analysed. It is observed that after SO pretreatment hydrogen activation/scrambling occurs at about 600 K and the reduction profile is characterised by a single reduction peak at 920 K. In contrast, a TPR/MO pretreatment of such SO sample leads to a facile hydrogen activation/scrambling and low temperature reduction profiles, the main reduction peak being observed at ca. 650 K. Using 1H2 and 2H2 as reducing agents, an isotopic effect is observed for the reduction processes with a peak occurring above 700 K, while equal activation energies are measured for those occurring below 700 K. This suggests that under the TPR conditions the surface reduction steps (activation of the reducing agent at the surface, surface reduction steps and water evolution) are not rate limiting for the low temperature reduction process, which is controlled by bulk diffusion, while some surface step (probably water formation) is limiting for the high temperature reduction. Studies by EPR on the nature of the oxygen species adsorbed on variously pretreated specimens and by FTIR of the methoxy species formed upon methanol adsorption on the same materials reveal that the main changes induced by high temperature calcination are related to segregation of ceria- and zirconia-type islands, with some surface enrichment in cerium probably occurring as well. Only subtle differences between SO and MO pretreated samples can be detected at either surface or bulk (XRD experiments) levels, in contrast with the significant modifications produced by the different pretreatments on the reduction characteristics of the sample.
1. Introduction
The so-called oxygen storage/release capacity (OSC) of a three-way catalyst (TWC) is the ability to attenuate the negative effects of rich/lean oscillations of exhaust gas composition. By maintaining a stoichiometric gas composition at the catalyst, the highest efficiency of the exhaust gas conversion is attained. OSC, which in the modern TWCs is achieved by adding a CeO2–ZrO2 mixed oxide component,1 is usually discussed in terms of the ability to regulate the oxygen partial pressure in the exhaust through the Ce3+/Ce4+ redox couple:
Both H2 and CO are present in the exhaust in a ratio 1 ∶ 3, which makes investigation of the reduction process, using either CO or H2 as reducing agent, of strong interest. Recently a number of investigations concerning reactions of CO with CeO2–ZrO2 mixed oxides which are related to the OSC property as described above appeared in the literature.2 CO oxidation was investigated under oscillating or constant feed-stream composition, or even in the absence of O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) , due to its relevance to the OSC property. We have investigated in detail the redox behaviour of these materials using H2 as reducing agent and showed that promotion of the redox behaviour of these systems could be related to structural modification of the CeO2 lattice by insertion of ZrO2, which makes the lattice oxygen highly mobile.3 Evidence from these
studies indicated that the reduction of CeO2, which is a slow process in comparison to CeO2 oxidation, could be rate limited by the mobility of the oxygen in the bulk. This proposal has been questioned, with the suggestion that processes other than those occurring in the bulk are rate limiting in the reduction process, even at low temperatures.2 There is a further point of interest in the redox-behaviour of the CeO2–ZrO2 mixed oxides, since it has been observed that high temperature reduction followed by mild oxidation leads to unusual improvements in the reduction behaviour at low temperatures.4 However, this behaviour is reversed by severe oxidation which moves the onset of the reduction processes to high temperatures.5 An understanding of the reduction mechanism at low temperatures, using H2 as reducing agent, is desirable since
it was shown recently that H2 can contribute efficiently to the OSC property, even at room temperature.6
, due to its relevance to the OSC property. We have investigated in detail the redox behaviour of these materials using H2 as reducing agent and showed that promotion of the redox behaviour of these systems could be related to structural modification of the CeO2 lattice by insertion of ZrO2, which makes the lattice oxygen highly mobile.3 Evidence from these
studies indicated that the reduction of CeO2, which is a slow process in comparison to CeO2 oxidation, could be rate limited by the mobility of the oxygen in the bulk. This proposal has been questioned, with the suggestion that processes other than those occurring in the bulk are rate limiting in the reduction process, even at low temperatures.2 There is a further point of interest in the redox-behaviour of the CeO2–ZrO2 mixed oxides, since it has been observed that high temperature reduction followed by mild oxidation leads to unusual improvements in the reduction behaviour at low temperatures.4 However, this behaviour is reversed by severe oxidation which moves the onset of the reduction processes to high temperatures.5 An understanding of the reduction mechanism at low temperatures, using H2 as reducing agent, is desirable since
it was shown recently that H2 can contribute efficiently to the OSC property, even at room temperature.6
In order to elucidate further these points, we report here temperature programmed reduction experiments carried out on a mixed oxide using both 1H2/Ar and 2H2/Ar mixtures as reducing agents. The changes in the surface state induced by the different pretreatments are probed by O2 adsorption, using the EPR technique. Recent studies showed the validity of this technique for evaluation of the type and intensity of superoxide radicals generated upon oxygen adsorption on the outgassed samples, whose presence is directly related to surface reducibility in the CeO2–ZrO2 mixed oxides.7 The studies on surface properties are complemented by infrared experiments using methanol as probe molecule. This method is based on monitoring the methoxy species formed upon methanol adsorption, which gives valuable information on the surface characteristics of this kind of material.8
A preliminary report on the TPR experiments described here appeared previously.9
2. Experimental
Ce0.5Zr0.5O2, with a BET surface area of 40 m2 g−1, was prepared by using a citrate route, as reported previously.10 Water was used as solvent. This sample, which was calcined at 773 K for 5 h, is designated as fresh. The sample was characterised by powder XRD using a Siemens Kristalloflex Mod.F Instrument with Ni-filtered Cu-Kα radiation.
The TPR/oxidation experiments were carried out as follows: the mixed oxide was pretreated in situ in flowing Ar at 873 K for 2 h, while pulsing pure O2
(100 µl) every 30 s. Such a cleaning pretreatment ensures a clean CeO2–ZrO2 surface, providing TPR profiles free from artifacts due to adsorbed species.11 Notice that water is desorbed by about 500 K. The sample was then cooled in Ar flow to room temperature (RT), the flow switched to either 5%
1H2 or 5%
2H2 in Ar and the temperature ramped at rates of 10–20 K min−1 up to 1273 K. The sample was held at this temperature for 15 min, then the gas was switched to Ar and the sample cooled to 700 K, whereupon it was re-oxidised by pulsing pure O2
(100 µl) every 30 s for 60 min. This treatment is described as mild oxidation (MO).
O2 uptake was also measured during MO by detecting the breakthrough point. The sample was then cooled to RT before a subsequent TPR run was carried out. In some experiments a severe oxidation (SO), i.e. in situ treatment in Ar at 1273 K for 2 h, while pulsing pure O2
(100 µl) every 30 s, was carried out before cooling the sample to RT. To ensure the reproducibility of the experimental conditions, all the pretreatments, post-treatments and the 1H2 TPR and 2H2 TPR were carried out in situ, on the same sample. H2 consumption was detected either by a conventional thermal conductivity detector (TPR/TCD) or by using a VG Sensorlab quadrupole mass spectrometer with PostSoft analysis software (TPR/MS). 25 mg of sample was employed in the TPR/TCD experiments, while 200 mg had to be employed for the TPR/MS. In the former case H2
(5%)
in N2 mixtures was also employed. High purity (99.999%) gases with no further purification were employed. However, due to ubiquitous water and long experimental times, we always detected the presence of 1H derived products in the TPR/MS experiments carried out with 2H2, as also evidenced by the presence of a peak at m/z=3.
EPR spectra were recorded, always at 77 K, with a Bruker ER 200 D spectrometer operating in the X-band and calibrated with a DPPH standard (g=2.0036). Portions of about 30 mg of sample were placed inside a quartz probe cell with greaseless stopcocks using a conventional high vacuum line (capable of maintaining a dynamic vacuum of ca. 0.006 N m−2) for the different treatments. Samples outgassed at Tv=773 K were analysed. Oxygen adsorption experiments were performed by adsorbing a dose of ca. 100 µmol of O2 per g of sample at 77 K. Then, after about 10 min contact, the sample was outgassed for 30 min at 77 K in order to minimise magnetic interactions with non-chemisorbed oxygen (this experiment will be hereafter referred to as oxygen adsorption at 77 K). The experiment was followed typically by warming to RT and finally outgassing at
RT for 15 min. In cases where intensity losses were noticed upon warming to RT, the evolution of the signals during the warming process was examined at different warming times.
Infrared spectra were recorded at RT with a Nicolet 5ZDX FTIR spectrometer. Self-supporting discs of the samples (containing 10–15 mg cm−2) were prepared and handled with a conventional static IR cell. Methanol adsorption (50 Torr) was performed at RT on samples previously outgassed at 773 K, followed by outgassing at RT for 15 min and finally outgassing at 373 K for 15 min.
3. Results and discussion
3.1. Bulk structural characteristics of the Ce0.5Zr0.5O2 solid solution
To avoid as far as possible artifacts due to phase separation under the redox-ageing conditions, a single phase Ce0.5Zr0.5O2 solid solution was prepared, which was characterised by XRD, as reported recently.12 Accordingly we will summarise only the most important features relevant to this study. Powder XRD patterns of the fresh, calcined (1273 K, 5 h) and redox-aged Ce0.5Zr0.5O2 showed no observable splitting of the (222) and (220) reflections, indicating the presence of a fluorite type cation lattice without detectable tetragonal distortion. By using a Rietveld profile fitting of the XRD patterns the cell parameter was calculated as 5.2969(8) Å and the particle size as 12 nm for the Ce0.5Zr0.5O2 calcined at 1273 K for 5 h. A redox-aged sample, i.e. subjected to TPR/MO and TPR/SO treatments as described in the Experimental section, was also analysed to check for structural modifications which could be induced by such an ageing procedure. It was observed that only negligible structural changes of the solid solution were induced by the redox-ageing: The cell parameter was calculated as 5.2816(8) Å and the particle size as 13 nm in the redox-aged sample. The Raman spectrum of the fresh Ce0.5Zr0.5O2 sample featured a strong broad band centred at 478 cm−1 with a shoulder at about 540 cm−1 and weak bands at 310 and 125 cm−1. This allowed attribution to a t′′ phase according to the classification proposed by Yashima et al.13 The intensity of the Raman bands increased somewhat after calcination at 1273 K, which is consistent with an increase in the particle size of the mixed oxide.14 In agreement with previous studies, upon redox-ageing the Raman spectrum appeared perturbed and the overall intensity of the Raman bands decreased.153.2. Spectroscopic studies of surface properties
In order to examine surface properties, analyses were carried out with EPR and FTIR, using respectively oxygen and methanol as probe molecules,7 on the fresh sample and on samples previously subjected to a SO treatment and to SO followed by TPR/MO treatment, indicated respectively as SO and MO samples. As shown below (Section 3.3), the SO and MO sample show distinct reduction behaviour, as most of the reduction occurs respectively above and below 700 K.Following an outgassing treatment at Tv=773 K, only a very weak symmetric signal at g=2.002 (signal A) is observed in the EPR spectrum of the fresh sample (data not reported). This signal appears typically on ceria-related materials after outgassing at relatively high Tv
(≥673 K) and can be most likely attributed to the generation of a small amount of particular anion vacancy defects at surface or subsurface positions in which single electrons are largely stabilised, according to their weak interaction with oxygen.16 Different signals appear after oxygen adsorption at 77 K on the fresh sample outgassed at Tv=773 K, the spectrum remaining unchanged upon subsequent warming to RT for 12 h. Higher spectral intensity and better resolution of the signals formed is however obtained after
subsequent outgassing at RT, which is likely attributed to the existence of magnetic interactions between the chemisorbed radicals and some residual non-chemisorbed oxygen species before this latter outgassing. The spectrum so obtained (Fig. 1(a)) is mainly formed by the overlapping of two signals, designated as OC1 and OZ, a minor signal designated as OC2 being also present. Table 1 shows a summary of the spectral characteristics of these oxygen-derived signals.
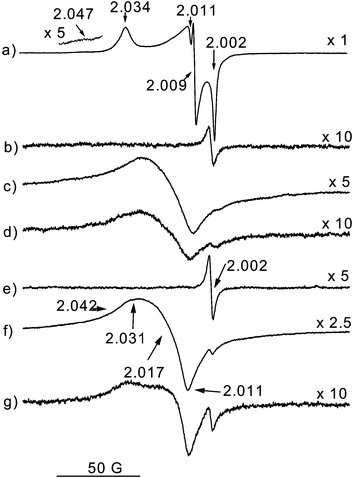 | ||
| Fig. 1 EPR spectra of the Ce0.5Zr0.5O2 specimens: (a) fresh sample after O2 adsorption at 77 K, warming to RT for 12 h and outgassing at RT. EPR spectra of sample MO: (b) outgassed at Tv=773 K; (c) subsequent O2 adsorption at 77 K; (d) subsequent warming for 1 min to RT. (e)–(g) The same as (b)–(d) for sample SO. | ||
| Signal | EPR parametersa | Assignment |
|---|---|---|
| a Axes attribution in the g-tensor follows criteria of previous studies.16 | ||
| OC1 |
g
‖=2.034, g⊥=2.011 |
O2−–Ce4+ formed on isolated oxygen vacancies |
| OC1′ |
g
z
=2.031, gx=2.017, gy=2.011 |
|
| OC2 |
g
‖=2.047–2.041, g⊥≈2.010 |
O2−–Ce4+ formed on associated oxygen vacancies |
| OZ |
g
z
=2.034, gy=2.009, gx=2.002 |
O2−–Zr4+ |
Certain similarities are observed in the nature and evolution of the EPR oxygen-derived signals detected on samples MO and SO during the various oxygen adsorption experiments. After outgassing at Tv=773 K, a signal A larger than that observed on the fresh sample (by factors of about 5 and 16 for samples MO and SO, respectively) is detected in both specimens, Fig. 1(b) and (e). The spectra observed for both samples after subsequent O2 adsorption at 77 K, Fig. 1(c) and (f), suggest the presence (apart from a small signal A, representing always an amount of spins below 0.1% of the total) of overlapped, relatively broadened signals with an overall intensity slightly higher for sample SO. (In relative terms, the intensities observed for the fresh, MO and SO samples, spectra in Fig. 1(a), (c)
and (f), are respectively 1, 0.79 and 1.05.) Careful analysis by computer simulation of these spectra is shown in Fig. 2; it allows us to verify that they are formed by a superposition of two signals OC1′ and OC2, sufficiently discernible via simulation in spite of their relatively larger linewidth and less accurately determined parameters; these latter are included in Table 1. The main difference between the spectra of the two samples concerns the relative intensities of these two signals: OC2/OC1′=3.5/1 for MO and 3/2 for SO (as obtained from doubly integrated intensities of the best-fit spectra: note that the change in signal amplitude ratio is somewhat different due to variations in line-widths). It is remarkable that no hint of signal OZ appears in the EPR experiments performed on these samples, in contrast to the fresh sample (Fig. 1(a)).
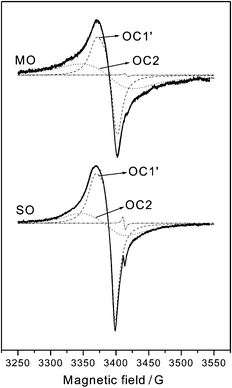 | ||
| Fig. 2 Experimental EPR spectra and computer simulation (individual components and total) of the spectra observed after O2 adsorption at 77 K on the indicated samples outgassed at Tv=773 K. | ||
For both samples MO and SO, and in contrast to the behaviour observed on the non-sintered fresh sample, a significant decrease and eventual disappearance of the signals is observed upon subsequent warming to RT (Fig. 3); this cannot be attributed to magnetic broadening effects of non-chemisorbed oxygen since no intensity recovery is observed after subsequent RT outgassing. It is noteworthy that similar behaviour, in terms of the type and shape of the signals present and their evolution during warming to RT, has been observed on a pre-sintered (by calcination under air at 1273 K for 12 h) pure ceria reference sample (experiments not shown), again contrasting with the behaviour of high surface area ceria.16 In general terms, it is observed that the intensity decrease upon warming to RT affects signal OC2 more quickly than signal OC1′, as inferred from analyses of the spectra shown in Fig. 1(d) and (g) compared to those observed after O2 adsorption at 77 K (Fig. 1(c) and (f)). Following the complete loss of these oxygen-derived signals after prolonged warming to RT, a second (and a third in the case of sample SO) oxygen dose was adsorbed at 77 K, followed by similar warming to RT. With these successive doses of oxygen, the spectra observed after adsorption at 77 K show similar signals as above but with progressively smaller overall intensity, while the signal intensity decrease observed upon subsequent warming to RT also becomes slower.
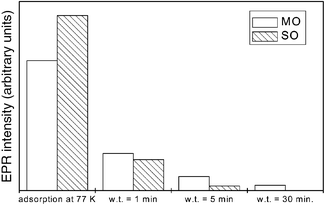 | ||
| Fig. 3 Evolution of the overall EPR integrated intensity of superoxide species in the O2 adsorption experiments performed on samples SO and MO; w.t. refers to warming time when warming to RT. | ||
Signals type OC1 (OC1 and OC1′) and OC2 are typical of O2− species adsorbed on coordinatively unsaturated Ce ions (cus-Ce, formally in the Ce4+ state).7 According to previous work, the differences observed between these signals with respect to their EPR parameters and to the thermal preconditioning required for their generation reveal the different nature of the oxygen adsorption sites where they are created, i.e. isolated anion vacancies and associated anion vacancies for signals type OC1 and OC2 respectively.16 These assignments have been supported by NO adsorption experiments showing that only the latter type of defect has NO reduction activity (yielding N2O) at low temperature;17 the presence of associated vacancy defects on single crystal ceria surfaces has also been evidenced experimentally
by scanning tunnelling microscopy (STM).18 Signal OZ, on the other hand, is attributed to O2−–Zr4+ species.7,19 Formation of the superoxide species requires prior generation of both surface oxygen vacancies, where oxygen may become adsorbed, and excess electrons, which may be transferred (to O2) from the sample surface. Considering that the reduction of cerium cations, as well as the generation of surface anion vacancies, can be achieved by the outgassing treatment, the formation of O2−–Ce4+ can be envisaged as process:

The formation of a large amount of O2−–Zr4+ species upon oxygen adsorption on the fresh sample, which contrasts with similar experiments on high surface area pure zirconia where very small amounts of such species appeared,11 evidences the promoting effect of cerium cations on the generation of vacancies in the coordinative environment of zirconium cations, suggesting that a mixed oxide-type morphology is present at the surface of that sample. This same characteristic was also detected in other Ce0.5Zr0.5O2 specimens of high homogeneity prepared by the microemulsion method,7 and can be considered demonstrative of the mixed oxide character of the sample surface. In contrast, the absence of O2−–Zr4+ in the experiments performed here on samples MO or SO indicates the lack of the influence of the
cerium component in enhancing vacancy formation on zirconium cations, suggesting that the mixed oxide character of the topmost surface is lost as a consequence of the high temperature thermal treatments. This might be due either to the absence of zirconium cations at the outermost surface layer (induced by a process of cerium segregation to the surface) or to the formation of zirconia islands of low reducibility coexisting with Zr-free ceria islands. The data show in any case that significant surface structure modifications are produced by the high temperature treatments. On the other hand, the decrease in the superoxide signals upon warming to RT in both samples MO and SO, which contrasts with the stability of such species in the fresh sample and reveals the higher facility of reoxidation of the surface vacancies created on these sintered materials, indicates that a higher overall reduction degree is achieved at their surface upon outgassing at Tv=773
K. Accordingly, considering the general scheme of oxygen reduction/surface reoxidation

FTIR spectra in the C–O bond stretching region obtained following methanol adsorption on the samples outgassed at Tv=773 K are shown in Fig. 4. The spectrum of the fresh sample is formed by bands at 1153, 1097 and 1058 cm−1
(Fig. 4(a)). A considerable intensity decrease is observed in the spectra of samples MO and SO, in correlation with their lower surface area (Fig. 4(b) and (c)). In both cases, the spectra show bands at 1157, 1102–1098 (with a shoulder at ca. 1120 cm−1), 1073–1070 cm−1 and 1027 cm−1. The main difference between the spectra of the two samples concerns the relative intensity of the band at ca. 1070 cm−1, which is considerably higher for sample MO.
 | ||
| Fig. 4 FTIR spectra following methanol adsorption at RT and outgassing at 373 K on the samples outgassed at Tv=773 K. (a) Fresh sample; (b) MO; (c) SO. | ||
According to previous studies, these bands are attributed to the ν(OC) stretching mode of methoxy species generated upon dissociative methanol adsorption.8 The frequencies of these bands are sensitive to the nature and redox state of the cations exposed at the surface where methoxy species are chemisorbed, and thus serve as a powerful tool for surface analysis.8
Following those studies, bands at 1157–1153 cm−1 and at 1102–1097 cm−1 are attributed to on-top methoxy species adsorbed, respectively, on Zr4+ and Ce4+ cations,8 while the shoulder at ca. 1120 cm−1 is attributed to on-top species adsorbed on Ce3+ cations.21 Bands at 1058 cm−1 and 1073–1070 cm−1 are attributed to doubly bridging methoxy species;8 for these complexes, the similarities in the frequencies observed for these bands in pure ceria and zirconia in comparison with the present ceria–zirconia mixed oxide samples make unreliable any attempt to determine the exact nature of the two cations presumably involved in the bonding, although the blue shift of the latter bands is a clear indication that the corresponding cerium cations are, on average, in a more reduced state.22 Finally, the band at 1027 cm−1 is related to triply bridging methoxy species.8 This band is absent in pure zirconia; over oxidised pure ceria, it appears at around 1013 cm−18 and shifts to higher frequency as the degree of reduction of ceria is increased.22
On the basis of these attributions, it is possible to conclude that one difference between the surface of the fresh sample and those of the MO or SO samples concerns the overall redox state attained upon outgassing at Tv=773 K in each case. While the spectrum of the fresh sample shows methoxy species adsorbed on oxidised cations, those of samples MO or SO present features of species adsorbed on both oxidised and reduced cerium cations. Correlation with the EPR results (Fig. 1–3) suggests that while both cus-Ce4+ and cus-Ce3+ ions are formed upon outgassing at Tv=773 K in every case, the former largely predominate on the fresh sample, indicating the higher surface reducibility under vacuum (in the conditions used here) of the low surface area samples MO or SO. This correlates well with the larger linewidth (which
may be due to a higher surface concentration of paramagnetic species, leading to magnetic dipolar broadening) and also with the lower stability (when warming to RT) of the EPR signals due to superoxide species formed at 77 K on the latter samples (Fig. 3), as discussed above; attainment of comparable intensities of superoxide species when comparing the fresh with MO or SO samples, in spite of the significant difference in specific surface, gives further correlation. On the other hand, the apparent red shift of the band due to on-top methoxy species adsorbed on Zr4+ which appears on the fresh sample (1153 cm−1) in comparison to that observed over high surface pure zirconia (1163 cm−18) can be attributed to the influence of nearby cerium cations, thus giving support to the above discussion on EPR results indicating that a mixed oxide
surface structure is actually present on that sample.
Concerning the surface structure of MO and SO samples, and recalling the above discussion on EPR results, the fact that the infrared spectra of these sintered specimens (Fig. 4) present bands due to on-top methoxy species adsorbed on Zr4+
(less blue-shifted from the pure zirconia position than those observed in the fresh sample) indicates that the absence of O2−–Zr4+ in the EPR spectrum of these samples (Fig. 2) must be interpreted rather as due to segregation of zirconia (or of a mixed oxide phase highly enriched in zirconium) in separate regions, forming at the surface of the samples islands which have little or no reducibility upon outgassing at Tv=773 K. Accordingly, the observation of O2−–Ce4+ species with characteristics similar
to those observed on pure ceria indicates that cerium appears at the surface as islands of ceria (or of a mixed oxide highly enriched in Ce). Anyway, some overall cerium enrichment at the surface seems also to be produced in the course of the thermal pretreatments undergone by these samples, since the intensity ratio (ICe/IZr) of on-top methoxy species adsorbed on cerium and zirconium cations is lower in the fresh sample than in the sintered specimens; note that such cerium-enrichment of surfaces with high temperature treatment is in line with results obtained by other research groups on similar samples.23,24 Beyond this result, the low S/N ratio of the FTIR spectra obtained here for samples SO and MO, together with the complexity of this spectral region, make it difficult to determine the nature of the adsorption sites producing bridging species, and thus prevents us from reaching more
definitive and detailed conclusions in this respect. As for the differences observed between MO and SO samples in the FTIR spectra, we can tentatively correlate the detection in sample MO of a higher proportion of the type of O2−–Ce4+ species related with associated vacancy centers (signal OC2, Fig. 2) with the higher amount of doubly bridging methoxy species detected in this specimen (Fig. 4). Further work is certainly required to clarify these points.
3.3. Effects of pretreatment on the TPR profiles
The effects of pretreatment on the reduction profiles of Ce0.5Zr0.5O2 obtained using 1H2 as reducing agent are reported in Fig. 5. Fresh Ce0.5Zr0.5O2 displays a single broad reduction feature centred at 850 K (trace a). This reduction pattern is consistent with previous observations indicating that TPR profiles of single phase CeO2–ZrO2 mixed oxides generally present a single reduction peak.20 The comparison of trace a with the baseline profile (non-horizontal due to buoyancy effects) included in Fig. 5 reveals however that there is a continuous broad H2 adsorption resulting in a significant tailing at the high temperature end of the reduction peak. The baseline was not recovered until isothermal reduction at 1273 K was carried out. As checked by using a mass spectrometer as detector, some water evolution corresponds to this H2 consumption. A possible explanation for this continuous water evolution/H2 consumption is that, apart from the reduction of the mixed oxide, deep sintering of the sample occurs, which leads to a collapse of the surface area from the initial value of 40 m2 g−1 to ca. 2 m2 g−1 after the first TPR. Such a process may well interfere with the reduction process, accounting for the continuous evolution. | ||
| Fig. 5 Effects of pretreatments on in situ temperature programmed reduction profiles (TPR/TCD) obtained for Ce0.5Zr0.5O2. Pretreatments: (a) fresh, (b) MO, (c) SO, (d) MO, (e) SO, (f) MO, (g) SO. Instrumental baseline is added under each trace. | ||
The TPR profile reported in trace b of Fig. 5, obtained on a sample which had been subjected only to a TPR run followed by MO treatment, just shows some displacement of the reduction peak to higher temperature compared to the fresh sample. This contrasts with a recent investigation of a Ce0.68Zr0.32O2 mixed oxide, where it was observed that the pretreatment conditions critically affect the redox behaviour.5 In that case, a TPR run followed by a MO pretreatment shifted the reduction process to low temperatures (below 800 K), while a SO pretreatment reversibly shifted the reduction process above 800 K; such observation was consistent with a previous study of this laboratory on the redox behaviour of a Ce0.5Zr0.5O2 mixed oxide15
(it should be noted, however, that the Ce0.5Zr0.5O2 mixed
oxide used in this latter study contained about 8% of a CeO2-rich minority phase, which could have affected the reduction behaviour25). In the study reported here, on the contrary, no shift of the reduction peaks to low temperature was observed, even for three consecutive TPR/MO sequences (data not shown). The reversible redox behaviour was however observed also on the present Ce0.5Zr0.5O2 mixed oxide when it was subjected to a SO/TPR/MO redox ageing (Fig. 5, trace d). After the sample was subjected to this kind of redox-ageing, the reduction process was split into two parts, most of the reduction occurring around 660 K while a minor peak remained at 900 K. A subsequent SO changed again the reduction profile, leading to a single reduction peak at 920 K; these changes were reversible, as evidenced by repeating alternately these treatments (Fig. 5(e)–(g)). It must be added that the presence of low temperature reduction processes as shown in traces d and f of Fig. 5 cannot be associated with incomplete desorption of water formed in the re-oxidation process, as this was checked separately by re-oxidising and adding some water at 700 K to a TPR/SO treated Ce0.5Zr0.5O2.
To summarise, the following three types of H2-reduction behaviour can be observed for Ce0.5Zr0.5O2, provided that single phase product is analysed: (i) a single, relatively broad high temperature (HT) reduction feature centred at approximately 850 K, which is characteristic of a fresh sample; (ii) a similar reduction profile as above, but shifted to somewhat higher temperature, which is characteristic of samples having been subjected to a SO-type calcination immediately before the TPR experiment (irrespective of their earlier history) as well as of samples which from the fresh state have been subjected to repetitive TPR/MO sequences, but never to a SO; and (iii) a predominant reduction peak at a low temperature (around 660 K, LT) followed by some residual reduction occurring around 900–950 K, which is observed for any sample after a TPR/MO sequence provided that
a SO-type treatment was applied in some previous moment. It may be added here that this latter SO-type treatment requires that the sample handling at high temperature is carried out on an oxidised sample; i.e. a MO-type oxidation followed by heating at 1273 K under inert gas atmosphere generates the SO-species as well. It thus appears that a SO pretreatment, i.e. high temperature treatment of an oxidised sample, performed either initially or during the redox-ageing, followed by a TPR/MO sequence is an essential prerequisite to induce the low temperature reduction behaviour. Indeed, once the sample has experienced the SO pretreatment the reduction behaviour becomes extremely sensitive to the experimental sequence: when the TPR is followed by MO treatment an easily reducible state is generated, and conversely, when a SO treatment is applied, a solid is obtained which is reducible only at high temperatures. For the sake of clarity, in the text we indicate the
species featuring the HT peak (case (ii) above) as SO pretreated, while the MO label designates a sample featuring the LT peak as it is generated from the SO sample by a TPR/MO treatment. Such MO and SO species easily and reversibly interconvert, as checked by alternating the TPR/MO and TPR/SO sequences at least 10 times. Notice that the present “reversible” redox behaviour is consistent with previous observations that also reported similar reversibility for samples of other compositions.26 It is also worth recalling that in both cases the samples had been subjected to high temperature calcination before use. These observations were recently confirmed by a comparative study of a series of high and low surface area CeO2–ZrO2 mixed oxides, the latter being generated from high surface area samples by calcination at 1173 K. These studies showed that the favourable effects of the TPR/MO
sequence on the reduction behaviour are strongly improved by pre-sintering of the CeO2–ZrO2 mixed oxide.27 However, such a high temperature oxidative pretreatment was not necessary to observe the promotional effect of TPR/MO when CeO2–ZrO2 mixed oxides containing CeO2 impurities were investigated (vide infra).15
3.4. Isotopic studies of the effects of pretreatments on the reactivity towards hydrogen
Hydrogen isotopic scrambling between the sample surface and gas phase hydrogen was detected during the TPR experiments by monitoring the m/z=3 signal (Fig. 6). Some signal at m/z=3 was always present in these traces, even in consecutive experiments carried out using 2H2 as a reducing agent, due to the unavoidable presence of traces of water in the feeding lines, as already indicated in the Experimental section. Anyway, the comparison of these results with those in Fig. 5, where a comparable sequence of pretreatments was applied, indicates that irrespective of the applied pretreatment hydrogen scrambling occurs at a temperature which is significantly lower than that of the corresponding reduction peak/peaks. This suggests that, independent of the pretreatment conditions, H2 is activated at
the surface before any so-called irreversible reduction28
(i.e. creation of oxygen vacancies) occurs. Even if one cannot discard the possibility that the activation of 2H2 for such a scrambling process could be of a different type from (or even unrelated to) that required for the reduction process, the reversible shifts of the temperature of the peak maximum for the scrambling process with the pretreatments, i.e. with the presence of MO or SO species, suggest that a correlation between the two phenomena does exist, at least in terms of the ability of the surface to activate H2. This is further reinforced by the fact that, as seen in Fig. 6, the scrambling behaviour of the species not subjected to the SO treatment is different from the SO-treated sample since it features a peak at approximately 470 K, which disappears after the SO treatment.
 | ||
| Fig. 6 Effects of pretreatments on hydrogen scrambling (m/z=3) during in situ temperature programmed reduction profiles (TPR/MS) obtained for Ce0.5Zr0.5O2. Pretreatments: (a) fresh, (b) MO, (c) MO, (d) SO, (e) MO, (f) SO, (g) MO. | ||
If the same reduction scheme which has been proposed for CeO229
(Scheme 1)† is considered also for Ce0.5Zr0.5O2, then these results suggest that reaction (1)
would not be a rate limiting step of the reduction process in any of the fresh, MO and SO states.
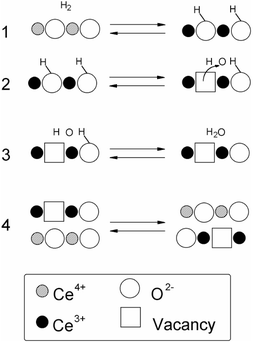 | ||
| Scheme 1 | ||
To further elucidate the reduction mechanism of Ce0.5Zr0.5O2 two series of experiments were carried out. First, a series of in situ TPR/TCD runs following the SO and MO pretreatments was carried out by using alternately 1H2 and 2H2 as reducing agent. This experiment was aimed at detecting isotopic effects on the reduction process under experimental conditions identical to those of Fig. 5. In fact the TPR profiles were hardly affected by experimental artifacts unless appropriate conditions were chosen.30 An analogous experiment, previously reported for CeO2, showed that the temperature of the TPR peak shifts reversibly by alternating 1H2 and 2H2 as reducing agent, evidencing the sensitivity of the TPR methodology to the isotopic effect.9 The TPR
profiles obtained in these experiments (data not shown) were quite consistent with those reported in Fig. 5; the corresponding TPR peak temperatures are reported in Table 2. The examination of the peak temperatures for the reduction feature occurring around 660 K, which is observed after the MO pretreatment, shows a remarkable agreement between the values measured when both isotopes are employed in the reducing agent. In contrast, for both MO and SO pretreated samples there is a small but significant and reproducible isotope influence on the temperature of the peak maxima observed around 900 K. In both cases, when 1H2 is employed as reducing agent the maximum reduction rate is observed at a lower temperature (7–12 K) than when using 2H2. Notice that all these experiments were performed in situ on the same sample, to avoid experimental artifacts.
| Pretreatment | Technique | Temperature of peak maxima Tm/K and activation energy Ea/kJ mol−1a | O2 uptake/ml g−1a | |||
|---|---|---|---|---|---|---|
| LT | HT | |||||
| T m | E a | T m | E a | |||
|
a Standard deviations: Activation energy ±3.5 kJ mol−1; O2 uptake 0.2 ml g−1.
|
||||||
| Fresh | TPR/1H2 | 850 | ||||
| SO | — | — | 914 | 90 | 17.5 | |
| MO | 659 | 39 | 896 | 91 | 17.4 | |
| MO | 663 | 896 | 17.6 | |||
| SO | TPR/2H2 | — | — | 921 | 134 | 17.4 |
| MO | 659 | 44 | 908 | 116 | 17.3 | |
| MO | 664 | 908 | 17.6 | |||
A second series of experiments was then carried out, varying the heating rate used in the TPR experiment. Monti and Baiker30 showed that by plotting ln(cMTM2/β)
versus 1/T, where cM, TM and β are respectively H2 concentration, temperature at peak maximum and heating rate, an apparent activation energy for the reduction process can be obtained. An example of the Arrhenius plots thus obtained is shown in Fig. 7, and the resulting apparent activation energies are given in Table 2. The insensitivity of the LT peak to the isotopic effect is confirmed by the apparent activation energies of about 40 kJ mol−1 measured in both cases, which are equal within experimental error. For comparison, it may be recalled
that apparent activation energies of about 50 kJ mol−1 were reported for CO oxidation over NM/CeO2 catalysts under conditions where oxygen migration from the support is believed to be rate limiting.31 In contrast, for the HT peak a much higher Ea is observed, which increases by about 25–35 kJ mol−1 when 2H2 is employed as reducing agent instead of 1H2.
 | ||
| Fig. 7 Arrhenius plot (ln(cMT2M/β) versus 1/T) for the (a) HT and (b) LT peaks in the TPR profiles of Ce0.5Zr0.5O2 subjected to a MO pretreatment. (1H2-TPR/TCD: filled symbols, 2H2-TPR/TCD: open symbols). | ||
These data strongly suggest that the rate determining step (RDS) of the reduction reaction is essentially different for the processes taking place within the LT and HT TPR peaks. In particular, the isotopic effect detected for the HT peak implies that the rate determining step in the latter includes forming and/or breaking of some bond(s) with hydrogen. As shown above, activation of the H2 molecule (step 1 in Scheme 1) occurs at temperatures clearly lower than those of the irreversible reduction; therefore this is unlikely to be the RDS. Rather, the rate limiting step for the reduction process above 700 K could be the formation of adsorbed water molecules, or also their desorption. The first alternative seems much more likely, in view of the large values of both the activation energy and its isotopic effect. In contrast, the absence of isotopic effect in the reduction occurring below 700 K, together with the relative similarity between the corresponding activation energy and that of ceria reduction by CO discussed above, is a strong indication that in this case the reduction process is limited by the rate of oxygen diffusion in the bulk of the mixed oxide. This is consistent with recent pulse CO/O2 experiments carried out over CeO2–ZrO2 which suggested the importance of bulk migration at low reaction temperatures.32
To corroborate these hypotheses, in situ TPR/MS experiments were carried out using both 1H2 and 2H2 as reducing agents; profiles of hydrogen consumption and water production were thus obtained during reduction of SO and MO pretreated samples. Despite the different experimental conditions employed in TPR/MS and TPR/TCD, which could affect the TPR profiles,33 a good agreement between the two techniques was achieved. Fig. 8 presents these data for the 2H2 case (together with the evolution of the m/z=3 signal, which reflects hydrogen scrambling); the water evolution curves obtained with the two isotopes for both samples are presented in Fig. 9, and the temperatures of the peak maxima are summarised in Table 3. The patterns
found for both MO and SO species indeed appear fairly consistent with those reported in Fig. 5, although there is now a shoulder at about 500–520 K preceding the main LT reduction peak in the MO pretreated sample, which can be noticed both as hydrogen consumption and water evolution, especially when using 2H2. This was not observed in the results of Fig. 5; there is no clear attribution of this difference, and it should be noted that, due to the higher amount of the sample which had to be used in the TPR/MS experiment, some perturbation of the TPR profiles could occur, particularly at low temperatures where water desorption or accumulation in transfer lines could interfere in the reduction process.
 | ||
| Fig. 8
2H2-TPR/MS profiles for the SO (1) and MO (2) pretreated Ce0.5Zr0.5O2. | ||
 | ||
| Fig. 9 Comparison of evolution of m/z=18 in the 2H2
(a) and 1H2
(b) TPR/MS profiles obtained on the SO (1) and MO (2) pretreated Ce0.5Zr0.5O2. | ||
| Pretreatment | m/za | Temperature of peak maxima/K | |||
|---|---|---|---|---|---|
| TPR/1H2-MS | TPR/2H2-MS | ||||
|
a As described in the Experimental section due to presence of traces of water, m/z=19, i.e. partially deuterated product was also detected.
|
|||||
| SO | 2 or 4 | 913 | 925 | ||
| 18 | 961 | 971 | |||
| 19 | 961 | 970 | |||
| 20 | — | 969 | |||
| MO | 2 or 4 | 660 | 895 | 660 | 910 |
| 18 | 678 | 910 | 675 | 938 | |
| 19 | 679 | 910 | 676 | 940 | |
| 20 | — | — | 678 | 940 | |
In any case, the water evolution curves shown here reflect the same result given by the TPD/TCD experiments, i.e. that the temperature of the maximum reduction rate is affected by the change of hydrogen isotope only for the HT reduction process, while the LT reduction is unaffected. Despite the small change in the molecular weight between deuterated and non-deuterated water, the reduction peak at 900 K is significantly affected by the change in the isotope, while much less difference is found for the LT process in the MO sample, except for the above-mentioned shoulder at 500 K (Fig. 9). In both peaks there is a visible shift between the temperatures of the maxima for hydrogen consumption and water desorption (15–20 K for the MO peak and 45–48 K for the SO sample), which is in agreement with the reduction scheme reported above, i.e. that the reduction process occurs via initial hydrogen activation (reversible reduction) followed by a slower water formation/desorption (irreversible reduction),34 although some contribution of experimental artifacts to this delay in water evolution cannot be excluded. In summary, these experiments suggest that under our experimental conditions water formation presumably limits the reduction rate in the process observed at high temperatures. Consistently, desorption of physically adsorbed water was completed by about 500 K, as observed in the initial cleaning pretreatment.11
3.5. Relationship between bulk diffusion and reduction rates
An estimation of the rate (or of any other related quantity) at which oxygen migrates from the bulk of the mixed oxide to the surface can be made and used to check the consistency of the reduction model given above. For such a calculation the bulk oxygen diffusion coefficient is needed. We are unaware of direct measurements of Do for a Ce0.5Zr0.5O2 mixed oxide. Values for it can, in principle, be calculated from electrical conductivity and transfer numbers data using the following expression:where Do (cm2 s−1) is the diffusion coefficient, σi (Ω−1 cm−1) the ionic conductivity, k the Boltzmann constant (J K−1), HF is the Haven ratio (0.78146 for a f.c.c. structure), T the temperature (K), Ci the concentration of charge carriers ((O atoms) cm−3) and Ze their charge (C). Such electrical property data have been reported by Chiodelli et al.35 for ceria–zirconia materials with several Ce : Zr ratios, prepared by ceramic methods; these solids calcined at high temperature should, in principle, be considered comparable to our SO samples. The values of Do calculated in this way are higher that those previously reported for CeO2: for example, for our Ce : Zr ratio we obtain Do
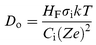 =2× 10−13 cm2 s−1 at 723 K, which is significantly higher than the values of 2 × 10−20 and 5 × 10−18 cm2 s−1 reported respectively
in ref. 36 and 37 for pure ceria. This is however consistent with recent studies of oxygen mobility in the bulk of CeO2–ZrO2 mixed oxides by Madier et al., who showed that Ce4+/Zr4+ isomorphous substitution enhances the exchange of oxygen in the bulk.38
=2× 10−13 cm2 s−1 at 723 K, which is significantly higher than the values of 2 × 10−20 and 5 × 10−18 cm2 s−1 reported respectively
in ref. 36 and 37 for pure ceria. This is however consistent with recent studies of oxygen mobility in the bulk of CeO2–ZrO2 mixed oxides by Madier et al., who showed that Ce4+/Zr4+ isomorphous substitution enhances the exchange of oxygen in the bulk.38
The relevance of the diffusion to the present TPR experiments can then be appreciated if the characteristic diffusion length (in the crystallites), defined as the average displacement due to diffusion in unit time interval (1 s) is determined (from the Do values) as a function of temperature (Fig. 10). Note that a discontinuity appears in this figure due to a change in activation energy and pre-exponential factor observed by Chiodelli et al.35 The mean diffusion path so computed equals the crystallite radius of our sintered Ce0.5Zr0.5O2 materials around 700 K, which is of the same order of magnitude as the temperature at which the maximum of the reduction rate (LT peak) occurs in the MO treated sample. In principle, these diffusion length values, computed from Chiodelli's data, are applicable to SO-type samples, but as an initial approximation we may take them as valid also for MO treated ones (more on this below). This result is thus already a first indication agreeing with the idea that in this latter material the overall solid reduction is achieved when diffusion allows the oxygen ions to travel from the bulk to the surface, even though the occurrence of grain boundary effects at high temperatures cannot be excluded.35 It is also compatible with a surface-related rate determining step in the case of SO samples, since at the temperature at which reduction of these is seen to occur, the anion diffusion in the bulk is much faster and does not limit the process.
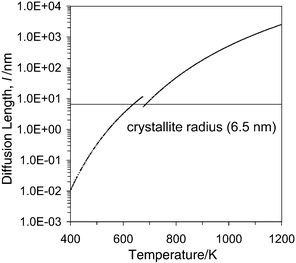 | ||
Fig. 10 Diffusion length (l)
versus temperature. Values calculated from the data reported in ref. 35 using the equation (diffusion time 1 s).
(diffusion time 1 s). | ||
To make the analysis more exact, one must take into account that in the TPR experiments oxygen atom migration occurs under an oxygen concentration gradient, due to the fact that the reduction proceeds from the surface to the bulk. Indeed, in the reduction occurring in MO samples within the LT peak, which is the one being discussed now, the surface reduction is presumed to occur (and be completed) faster than that of the bulk, as suggested by the joint consideration of the hydrogen scrambling experiments reported above (which concern the activation of hydrogen) and of the lack of isotopic effect in this main reduction peak (which implies that the formation of water, and consequently of surface anion vacancies, is not rate determining). Thus one may assume, as a first approximation, that the external surface is completely reduced during this LT reduction, i.e. at the surface region the oxygen ion concentration would be 87.5% of the maximum.
Upon application of Fick's law of diffusion the oxygen flux N
(molecules cm−2 s−1) to the surface can be calculated:2
 =2 in Fig. 11), the experimental rate observed at the beginning of the reduction process is much higher than the calculated flux. For the sake of comparison, Fig. 11 reports some additional curves (lower x values) which represent intermediate degrees of reduction of the bulk; qualitative analysis reveals differences which would again imply that the observed flux is higher than predicted from the diffusion model used. This might indicate that the diffusion coefficient in the MO-treated sample is higher than that of the SO-type solids (as in the work by Chiodelli et al.); however, this conclusion is not firm
since DO can change when varying the amount of oxygen vacancies in the bulk.39 Finally, it is important to note that the rate of diffusion of the oxygen atoms to the surface depends on the surface area of the sample; the higher the surface area, the lower r and consequently the higher the flux. This could easily contribute to the sensitivity of the reduction behaviour of the CeO2–ZrO2 mixed oxides to the textural properties.
=2 in Fig. 11), the experimental rate observed at the beginning of the reduction process is much higher than the calculated flux. For the sake of comparison, Fig. 11 reports some additional curves (lower x values) which represent intermediate degrees of reduction of the bulk; qualitative analysis reveals differences which would again imply that the observed flux is higher than predicted from the diffusion model used. This might indicate that the diffusion coefficient in the MO-treated sample is higher than that of the SO-type solids (as in the work by Chiodelli et al.); however, this conclusion is not firm
since DO can change when varying the amount of oxygen vacancies in the bulk.39 Finally, it is important to note that the rate of diffusion of the oxygen atoms to the surface depends on the surface area of the sample; the higher the surface area, the lower r and consequently the higher the flux. This could easily contribute to the sensitivity of the reduction behaviour of the CeO2–ZrO2 mixed oxides to the textural properties.
 | ||
| Fig. 11 Comparison of experimental reduction rate measured on Ce0.5Zr0.5O2 subjected to the SO/TPR/MO pretreatment with the calculated oxygen flux to the surface (2 m2 g−1); x represents the degree of reduction in the bulk of Ce0.5Zr0.5Ox. | ||
3.6. Implications of the data for models of ceria–zirconia reduction
Two main points arise from the preceding results and discussions. First, the important and reversible modification in redox behaviour observed when cycling between MO and SO states requires in these samples a previous SO-type high temperature oxidation. The spectroscopic measurements suggest that the modification induced by this treatment may have, as a key aspect, a change in surface structure in which pure ceria-like islands appear (the possible simultaneous existence of some pure zirconia-like islands being not detrimental in this); the fact that, in other studies using samples which contained a CeO2 impurity, such previous SO treatment was not necessary to obtain the reversible behaviour agrees with this.Secondly, the question arises: what is the reason for this reversible effect? The TPR data, with the observed isotopic effects included, indicate clearly that in SO samples the reduction of the surface, and specifically some step involving breaking or formation of O–H bonds, is difficult and constitutes the RDS for the reduction of the bulk by H2, which consequently only occurs at temperatures around 900 K; but in MO samples the reduction of the surface is made much easier, to such an extent that it can occur at temperatures below 550 K. A consequence is that the overall reduction rate becomes limited by the oxygen diffusion in the bulk, which begins to be fast enough around 600–700 K. It must be noted here that, within this scheme, the data reported in this work do not clarify whether in SO-type samples the oxygen diffusion rate in the bulk is different from that in MO-type samples, although the study summarised in Fig. 11
suggests that such diffusion could be somewhat faster in the latter. The reason for this large change in surface behaviour, however, remains unclear. The spectroscopic data do not show large changes in surface composition, although rearrangements of the surface cations in their mutual lateral disposition might occur, as hinted by the FTIR spectra. The differences in surface structure would thus be of some subtle nature. Certainly the EPR data would imply that the reducibility by vacuum treatments is not very different in the MO and SO states; if anything, the surface of the latter would be more reducible, in apparent contradiction with the TPR results—but these latter use H2 as reducing agent, which is a different story since the isotopic study shows that the difference lies just in reaction steps involving O–H bonds.
Although the situation remains still unresolved in this respect, it is worth noting that there might be some connection between the redox behaviour observed here and the results of neutron diffraction studies reported by Mamontov et al.40 These authors detected, in reoxidised ceria–zirconia materials, oxygen atoms located in rather substantial amounts in interstitial positions of the structure (in octahedral interstices of the f.c.c. cation lattice), while part of the normal anion sites (the tetrahedral ones) remained unoccupied. The same observation was reported also by Thomson et al.41 Such interstitial oxygens accompanied by vacancies in the regular lattice sites (i.e. Frenkel defects) could be detected also by Mamontov et al.40 in pure ceria; in this latter oxide the interstitials disappeared, filling the regular sites,
upon calcination above ca. 900 K, but for their ceria–zirconia material (having atomic ratio Ce : Zr=2 : 1) their amount remained unchanged after treatment up to 1050 K (the highest temperature used there). Those authors pointed out that this interstitial oxygen species could be more mobile and easily removable than the regular lattice ones, so that they could be responsible for the good oxygen buffering properties of ceria–zirconia. It is tempting to hypothesise that such interstitials could be present in our MO-treated specimens but be absent in SO-type samples due to their recombination with the anion lattice site vacancies upon calcination at some temperature in the 1050–1250 K range (in a similar way as shown to occur in ref. 40 for pure ceria at lower temperatures); and that these interstitials could then be responsible for the LT peak detected in TPR
experiments on the MO samples. Of course, if this were so, among those interstitial species only those present at the surface would play an important role, since the main large difference between both types of sample is related, according to the discussion above, to the different ease of water formation at the surface during reduction. It is not unlikely that such species could have a basicity significantly different from that of regular anion sites, strongly modifying the energetics and kinetics of the water formation steps. The verification of such a hypothesis would require other types of experiments, able to discriminate the properties of any such unusual anion species at the surface.
4. Conclusions
The present work confirms that the reduction behaviour of Ce0.5Zr0.5O2 solid solution is strongly affected by pretreatments. A relatively stable, single-peak reduction profile can be observed on a high surface area, single-phase product of this stoichiometry. A high temperature oxidative treatment, leading to strong sintering, is necessary to modify this behaviour, generating a system whose reduction behaviour becomes highly sensitive to the pretreatment. As revealed by the spectroscopic study of O2 and methanol adsorption, the surface structure and reactivity are strongly modified by such oxidative pretreatment: pure ceria-like surface islands appear and higher amounts of surface oxygen vacancies are generated by vacuum treatment which are more easily reoxidised than on the initial fresh sample. However, according to these techniques the thermal cycling treatments which modify strongly and reversibly the redox ability of the high temperature preoxidised material do not lead to large structural changes at the surface. Use of 1H2 and 2H2 as reducing agent under the TPR conditions has allowed us to elucidate important features of the reduction mechanism of these CeO2–ZrO2 mixed oxides. It appears that the low-temperature reduction behaviour of the CeO2–ZrO2 mixed oxides observed in samples reoxidised at low temperature have their rate controlled by the properties of the bulk of the oxide, the surface processes being faster in comparison. In contrast, the reduction features occurring at higher temperatures (the only ones present in samples reoxidised at high temperature) are controlled by surface processes, possibly related to water evolution, which are now slower than anion diffusion in the bulk.Acknowledgements
Helpful discussions with Prof. Alessandro Trovarelli (University of Udine), Dr. Gabriele Balducci and Dr. Roberta Di Monte (University of Trieste) are gratefully acknowledged. Dr. Roberta Di Monte is also acknowledged for the XRD measurements. CICYT (project Nr. MAT2000-1467), University of Trieste, CNR (Roma) Programmi Finalizzati “Materiali Speciali per Tecnologie Avanzate II, Contract n. 97.00896.34 and “Regione Friuli Venezia-Giulia, Fondo regionale per la ricerca” are gratefully acknowledged for financial support. A. B. H. and A. M.-A. wish to thank the Comunidad de Madrid for grants under which this work has been carried out.References
- J. Kaspar, M. Graziani and P. Fornasiero, in Handbook on the Physics and Chemistry of Rare Earths: The Role of Rare Earths in Catalysis, ed. K. A. Gschneidner, Jr. and L. Eyring, Elsevier, Amsterdam, 2000, ch. 184, pp. 159–267. Search PubMed.
- C. E. Hori, H. Permana, K. Y. S. Ng, A. Brenner, K. More, K. M. Rahmoeller and D. N. Belton, Appl. Catal. B, 1998, 16, 105 CrossRef CAS.
- P. Fornasiero, R. Di Monte, G. Ranga Rao, J. Kaspar, S. Meriani, A. Trovarelli and M. Graziani, J. Catal., 1995, 151, 168 CrossRef CAS.
- G. Balducci, P. Fornasiero, R. Di Monte, J. Kaspar, S. Meriani and M. Graziani, Catal. Lett., 1995, 33, 193 Search PubMed.
- R. T. Baker, S. Bernal, G. Blanco, A. M. Cordon, J. M. Pintado, J. M. Rodriguez-Izquierdo, F. Fally and V. Perrichon, Chem. Commun., 1999, 149 RSC.
- N. Hickey, P. Fornasiero, J. Kaspar, M. Graziani, G. Blanco and S. Bernal, Chem. Commun., 2000, 357 RSC.
- A. Martinez-Arias, M. Fernandez-Garcia, C. Belver, J. C. Conesa and J. Soria, Catal. Lett., 2000, 65, 197 Search PubMed.
- G. Colon, M. Pijolat, F. Valdivieso, H. Vidal, J. Kaspar, E. Finocchio, M. Daturi, C. Binet, J. C. Lavalley, R. T. Baker and S. Bernal, J. Chem. Soc., Faraday Trans., 1998, 94, 3717 RSC.
- P. Fornasiero, J. Kaspar and M. Graziani, Appl. Catal. B, 1999, 22, L11 CrossRef CAS.
- P. Vidmar, P. Fornasiero, J. Kaspar, G. Gubitosa and M. Graziani, J. Catal., 1997, 171, 160 CrossRef CAS.
- M. Daturi, C. Binet, J. C. Lavalley, H. Vidal, J. Kaspar, M. Graziani and G. Blanchard, J. Chim. Phys., 1998, 95, 2048 Search PubMed.
- P. Fornasiero, R. Di Monte, J. Kaspar, T. Montini and M. Graziani, Stud. Surf. Sci. Catal., 2000, 130, 1355 Search PubMed.
- M. Yashima, H. Arashi, M. Kakihana and M. Yoshimura, J. Am. Ceram. Soc., 1994, 77, 1067 Search PubMed.
- G. W. Graham, W. H. Weber, C. R. Peters and R. K. Usmen, J. Catal., 1991, 130, 310 CrossRef CAS.
- P. Fornasiero, G. Balducci, R. Di Monte, J. Kaspar, V. Sergo, G. Gubitosa, A. Ferrero and M. Graziani, J. Catal., 1996, 164, 173 CrossRef CAS.
- J. Soria, A. Martinez-Arias and J. C. Conesa, J. Chem. Soc., Faraday Trans., 1995, 91, 1669 RSC.
- A. Martinez-Arias, J. Soria, J. C. Conesa, X. L. Seoane, A. Arcoya and R. Cataluna, J. Chem. Soc., Faraday Trans., 1995, 91, 1679 RSC.
- H. Nöremberg and G. A. D. Briggs, Phys. Rev. Lett., 1997, 79, 4222 CrossRef.
- M. Anpo, M. Che, B. Fubini, E. Garrone, E. Giamello and M. C. Paganini, Top. Catal., 1999, 8, 189 Search PubMed.
- J. Kaspar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285 CrossRef CAS.
- F. Fally, V. Perrichon, H. Vidal, J. Kaspar, G. Blanco, J. M. Pintado, S. Bernal, G. Colon, M. Daturi and J. C. Lavalley, Catal. Today, 2000, 59, 373 CrossRef CAS.
- M. Daturi, E. Finocchio, C. Binet, J. C. Lavalley, F. Fally, V. Perrichon, H. Vidal, N. Hickey and J. Kaspar, J. Phys. Chem. B, 2000, 104, 9186 CrossRef CAS.
- V. Sergo and D. R. Clarke, J. Am. Ceram. Soc., 1995, 78, 641 Search PubMed.
- G. W. Graham, C. L. Roe, L. P. Haack and A. M. Straccia, J. Vac. Sci. Technol. A, 2000, 18, 1093 CrossRef CAS.
- T. Egami, W. Dmowski and R. Brezny, Soc. Automot. Eng., [Spec. Publ.] SP, 970461, 1997. Search PubMed.
- S. Otsuka-Yao-Matsuo, T. Omata, N. Izu and H. Kishimoto, J. Solid. State. Chem., 1998, 138, 47 CrossRef CAS.
- H. Vidal, J. Kaspar, M. Pijolat, G. Colon, S. Bernal, A. M. Cordon, V. Perrichon and F. Fally, Appl. Catal. B, 2000, 27, 49 CrossRef CAS.
- S. Bernal, J. J. Calvino, G. A. Cifredo, J. M. Rodriguez-Izquierdo, V. Perrichon and A. Laachir, J. Catal., 1992, 137, 1 CrossRef CAS.
- J. El Fallah, S. Boujana, H. Dexpert, A. Kiennemann, J. Majerus, O. Touret, F. Villain and F. Le Normand, J. Phys. Chem., 1994, 98, 5522 CrossRef CAS.
- D. A. M. Monti and A. Baiker, J. Catal., 1983, 83, 323 CrossRef CAS.
- T. Bunluesin, E. S. Putna and R. J. Gorte, Catal. Lett., 1996, 41, 1 Search PubMed.
- M. Boaro, C. de Leitenburg, G. Dolcetti and A. Trovarelli, J. Catal., 2000, 193, 338 CrossRef CAS.
- G. Fierro, M. LoJacono, M. Inversi, P. Porta, R. Lavecchia and F. Cioci, J. Catal., 1994, 148, 709 CrossRef CAS.
- S. Bernal, J. J. Calvino, G. A. Cifredo, J. M. Gatica, J. A. Perez-Omil and J. M. Pintado, J. Chem. Soc., Faraday Trans., 1993, 89, 3499 RSC.
- G. Chiodelli, G. Flor and M. Scagliotti, Solid State Ionics, 1996, 91, 109 Search PubMed.
- M. Kamiya, E. Shimada and Y. Ikuma, J. Ceramic. Soc. Jpn., 1998, 106, 1023 Search PubMed.
- D. Martin and D. Duprez, J. Phys. Chem., 1996, 100, 9429 CrossRef CAS.
- Y. Madier, C. Descorme, A. M. LeGovic and D. Duprez, J. Phys. Chem. B, 1999, 103, 10999 CrossRef CAS.
- B. C. H. Steele and J. M. Floyd, Proc. Br. Ceram. Soc., 1971, 72, 55 Search PubMed.
- E. Mamontov, T. Egami, R. Brezny, M. Koranne and S. Tyagi, J. Phys. Chem. B, 2000, 104, 11110 CrossRef CAS.
- J. B. Thomson, A. R. Amstrong and P. G. Bruce, J. Solid State Chem., 1999, 148, 56 CrossRef CAS.
Footnote |
| † The reaction mechanism shown in Scheme 1 follows that reported in ref. 29, however, it should be noted that the removal of water in the dehydroxylation of oxide surfaces (both in reductions and in other processes) is considered to be initiated by transfer of a proton from an OH group (more or less acidic) to another OH (more or less basic) to form cation-coordinated (adsorbed) H2O, this latter subsequently desorbing from the cation(s) into the gas phase, rather than a single step process. |
| This journal is © the Owner Societies 2002 |