Spontaneous doping and magnetic properties of polyacetylene and polypropyne synthesized in situ in Ni-exchanged mordenite and mesoporous MCM-41
María S.
Galletero
a,
Mercedes
Alvaro
a,
Hermenegildo
García
*a,
Carlos J.
Gómez-García
b and
Alexander K.
Lay
c
aInstituto de Tecnología Química CSIC-UPV and Department of Chemistry, Universidad Politécnica de Valencia, Apartado 46071-Valencia, Spain
bInstituto de Ciencia Molecular (ICMoI), Departamento de Química Inorgánica, Universitat de València, Dr. Moliner 50, 46100 Burjassot (Valencia), Spain
cChemistry Department, University of Reading, Whiteknights, P.O. Box 224, Berkshire, Reading, UK RG6 6AD
First published on 5th December 2001
Abstract
Upon polymerisation of acetylene and propyne inside the channels of Ni2+-exchanged mordenite and mesoporous MCM-41 spontaneous doping and formation of antiferromagnetic NiO clusters are observed to various extents. The population of polarons present in the final polymer/zeolite composite: (i) is higher for polyacetylene than for polypropyne; (ii) increases with polymerisation temperature in the range 100 to 335![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C; (iii) increases with the C content; and (iv) is higher for mordenite than for MCM-41 under the same conditions. The use of Ni(0),H+-mordenite for polymer formation enhances the polaron population of the resulting polymer by over one order of magnitude. Doped polyacetylene encapsulated in mordenite or MCM-41 was found to be persistent for periods of over three months after its preparation.
°C; (iii) increases with the C content; and (iv) is higher for mordenite than for MCM-41 under the same conditions. The use of Ni(0),H+-mordenite for polymer formation enhances the polaron population of the resulting polymer by over one order of magnitude. Doped polyacetylene encapsulated in mordenite or MCM-41 was found to be persistent for periods of over three months after its preparation.
Introduction
The preparation of conducting polymers inside the channels of zeolites is a promising method to obtain hybrid materials with enhanced mechanical and chemical performance over that of the pure conducting polymers.1,2 The case of polyacetylene is particularly interesting. Polyacetylene is the simplest organic conducting polymer and has the highest intrinsic conductivity and charge storage capacity per unit mass.3 However, polyacetylene is also one of the less processable polymers. In addition, when suitably doped, polyacetylene is very prone to undergo oxidative degradation by reaction with molecular oxygen, limiting its real applicability. Preparation of polyacetylene and polypropylene by direct alkyne polymerisation in acid H-ZSM-5 and H-mordenite4,5 or basic Cs+-exchanged mordenite6,7 has been previously reported but only low polyacetylene loading (<10 wt.%) was achieved. Furthermore, the possibility that polymerisation could be accompanied by spontaneous polaron formation has not been addressed. Encapsulation of polarons inside zeolites could find application in antistatic coatings and charge storage devices.More recently, we have observed that a much higher loading of polyacetylene, filling a large volume of the free zeolite voids, can be obtained by using Ni2+-exchanged porous aluminosilicates and performing the direct polymerisation of alkynes at temperatures ranging from 150 to 300°C.8 Thus, a loading of over 25 wt.% polyacetylene was obtained by polymerising acetylene within mesoporous Ni2+-exchanged MCM-41. Analogously, when the same polymerisation procedure is applied to Ni2+-exchanged zeolites such as mordenite, Y and beta zeolites, relatively high loading is also achieved. The structure of the polymer was unambiguously characterised by diffuse reflectance UV–Vis, FT Raman and MAS 13C-NMR. It was observed that polymerisation is accompanied by the formation of graphite fibres when the initial exothermic polymerisation peak is not properly
controlled.8
A well-established property of zeolites is their ability to spontaneously generate radical cations upon incorporation of electron-rich organic guests. The types of molecule whose radical cations have been detected in zeolites include amines,9–11 sulfur heterocycles,12–16 polycyclic aromatic hydrocarbons17 and α,ω-diphenylpolyenes.18–20 In fact, generation of minute but detectable amounts of acetylene radical cation by adsorption of acetylene on H-mordenite has already been reported.21 In view of these precedents, it is of interest to determine whether or not in situ polymerisation of acetylene and propyne within zeolites or mesoporous aluminosilicate is accompanied by spontaneous doping of the resulting polymer included inside the channels. This spontaneous doping, leading to polaron formation during the polymerisation process, is very likely in view of the fact that electron-rich organic molecules of similar oxidation potential to that reported for the onset of polyacetylene electrochemical oxidation (∼1 V vs. SCE)22 undergo, at low loading, a complete transformation to their radical cations in acid zeolites. In fact, the reported oxidation capability of zeolites is about 1.5 V.18 Moreover, the possibility of an enhanced doping is of particular importance considering the potential application of these doped materials as electrodes in lithium batteries.
Herein we report EPR spectra and magnetic susceptibility measurements that prove that such spontaneous doping of polyacetylene and polypropyne indeed occurs. The extent of the doping increases with the polymerisation temperature and carbon content. Unexpected formation of NiO clusters during the polymerisation process was also observed. Over one order of magnitude enhancement of the polaron density was achieved by previous reduction of Ni2+ to Ni(0).
Results and discussion
Polymerisation of acetylene within Ni2+-exchanged mordenite (Si/Al 10) and Al/MCM-41 (Si/Al 13) was accomplished by admitting, at atmospheric pressure, purified acetylene gas onto thermally dehydrated solids at controlled reaction temperatures. The main analytical data of the samples prepared are summarised in Table 1. Diffuse reflectance UV–Vis spectra of the hybrid solids after polymerisation indicate that all the samples contain polyalkyne, except sample 11. Some differences in the spectra were observed, related to the presence of graphite. Fig. 1 illustrates a selection of the diffuse reflectance UV–Vis spectra of the samples. Raman spectroscopy also gives results in agreement with the diffuse reflectance UV–Vis spectra.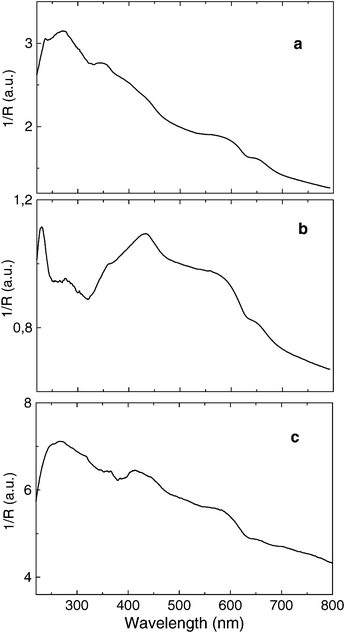 | ||
| Fig. 1 Diffuse reflectance UV–Vis spectra of samples 1 (a), 3 (b) and 6 (c). | ||
| Sample number | Polymer | Host | T/°C | Carbon content (%) | C/H ratio |
|---|---|---|---|---|---|
| a No polymer was observed by diffuse reflectance UV–Vis and Raman spectroscopy. | |||||
| 1 | PA | Ni2+-mor | 300 | 11.58 | 12.52 |
| 2 | PA | Ni2+-mor | 220 | 10.21 | 9.63 |
| 3 | PA | Ni(0)-mor | 100 | 0.99 | 0.87 |
| 4 | PP | Ni(0)-mor | 100 | 6.47 | 4.75 |
| 5 | PA | Ni(0)-mor | 285–335 | 28.16 | 24.70 |
| 6 | PA | Ni(0)-mor | 250 | 3.72 | 3.29 |
| 7 | PP | Ni(0)-mor | 290 | 6.46 | 4.71 |
| 8 | PA | Ni2+-Al/MCM-41 | 75–100 | 6.47 | 2.72 |
| 9 | PA | Ni2+-Al/MCM-41 | 150–165 | 25.38 | 12.94 |
| 10 | PA | Ni2+-Al/MCM-41 | 225 | 32.51 | 18.16 |
| 11 a | PP | Ni2+-Al/MCM-41 | 23 | 9.54 | 3.48 |
The carbon content can be taken as a measure of the polymer loading. As can be seen in Table 1, it increases with the polymerisation temperature and under similar conditions is higher for Al/MCM-41 than for mordenite. This reflects the higher pore volume of MCM-41 compared to mordenite. On the other hand a C/H ratio higher than 12 or 9 for polyacetylene and polypropyne, respectively, indicates an excess of carbon, related to the presence of graphite.8 This is the case for samples 1, 5 and 10.
Since the present study is focused on the presence of polarons in the polyacetylene/aluminosilicate hybrid solids, it is of interest to study the possibility of promoting chemical doping during polymerisation. With this aim, we wanted to know whether Ni(0) atoms are active sites for acetylene polymerisation and whether these reduced Ni(0) clusters can be effective in the doping of the polyacetylene. In this way, treatment of alkyne with Ni-exchanged aluminosilicates could not only result in polymerisation but also in a significant enhancement of the doping of the polymer which would be consequently charged and ready for use as an electrode. The stability of encapsulated polarons to the ambient is also of interest. To obtain Ni(0) clusters inside the zeolite micropores, a sample of Ni2+-exchanged mordenite was treated for several hours with a flow of hydrogen gas from room temperature up to 750°C using a smooth temperature ramp. The progress of the Ni2+ reduction was followed by monitoring the hydrogen uptake using a calibrated thermal conductivity detector. The resulting thermoprogrammed reduction plot is shown in Fig. 2. The plot has two regions where similar hydrogen uptake occurs. This can be easily rationalized as indicating a two-step reduction from Ni(II) to Ni(0). After the reduction, the colour of the mordenite changes from white to black and accordingly the resulting diffuse reflectance UV–Vis spectrum of the resulting Ni(0),H+-mordenite exhibits a continuous absorption throughout all the wavelength range, with an increasing slope towards the short wavelength region.
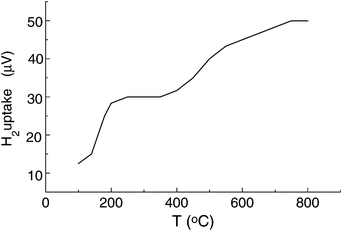 | ||
| Fig. 2 Thermoprogrammed reduction of Ni2+-Y by hydrogen flow. | ||
Influence of the polymerisation temperature
It has been found that the reaction temperature and the occurrence or not of an intense, initial exothermic peak during the polymerisation are among the most important factors that influence the quantity and quality (i.e. absence or presence of graphite) of the polyacetylene formed.8 We observed that the initial exothermic peak is related to the pH at which the Ni2+ ions were ion exchanged in the solid. Thus, samples 1 and 5 in which Ni2+ ion exchange was carried out at neutral pH, exhibit in the initial polymerisation stages a spontaneous intense exothermic peak. This exothermic process could be correlated with the thermal decomposition of acetylene to carbon and hydrogen. In addition to the formation of graphite, acetylene polymerisation also occurs in all samples except sample 11 in which no polypropyne was observed. Excess of C, indicating the formation of graphite, is, however, not observed for other Ni2+ zeolites for which the Na+-to-Ni2+ exchange is conducted at pH 5. The formation of graphite as a function of the pH for the Ni2+ ion exchange can be attributed to the negative influence on acetylene polymerisation of nickel oxide clusters. NiO clusters are formed from nickel hydroxides introduced during the ion exchange at neutral pH, according to the simplified hydrolysis equation (eqn. (1)). Thus, high-quality, graphite-free polyacetylene/zeolite solids such as samples 2, 7 and 9 require Ni-zeolites lacking NiO clusters or external nickel aggregates and a careful control of the polymerisation temperature.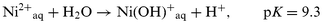 | (1) |
Besides the occurrence of an initial exothermic peak, the polymerisation temperature also controls the amount of polymer formed. We carried out the polymerisation of acetylene and propyne at different reaction temperatures. An increase in the polymerisation temperature clearly increases the carbon content of the solid (compare samples 3 and 6 or 8, 9 and 10 in Table 1). This allows us to correlate the polymerisation temperature and sample carbon content with the EPR spectra and magnetic properties.
EPR spectra and magnetic measurements
Typically the EPR spectra of samples 1–10 recorded at r.t. consist of two signals (see Fig. 3). Table 2 lists the relative intensity of these two signals measured at 300 and 4.2 K. One of the signals was extremely broad (ΔH∼2000 G) and centred around 3000 G. This signal cannot correspond to an organic polymer. Decreasing the temperature during the recording of the EPR spectra, reduces dramatically the intensity of this broad background signal, indicating that the species exhibits antiferromagnetic coupling. Based on these observations and also on the measurements using a superconducting quantum interface device (SQUID)
(see below), we attribute this broad signal to NiO clusters. Formation of these NiO clusters is more abundant in mordenite than in MCM-41 and does not depend exclusively on the thermal treatment of the aluminosilicate.
In fact, all the samples were thermally dehydrated at 300°C prior to the polymerisation and some of them contain lesser amounts of NiO (see Table 2). A likely possibility would be that the formation of NiO clusters occurs concurrently with the formation of graphite. It seems that the exothermicity of the graphite producing process (probably reaching very high local temperatures) is responsible for the agglomeration and subsequent migration of these NiO particles, particularly in the narrower channels of mordenite.
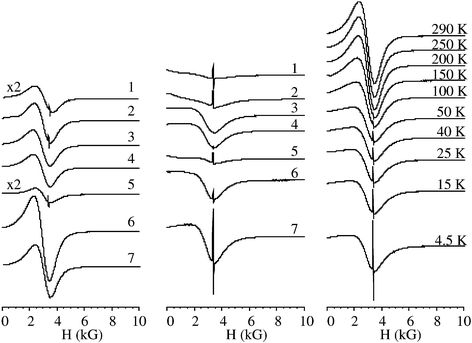 | ||
| Fig. 3 EPR spectra at room temperature (left) and 4.2 K (center) of mordenite samples. Right: Thermal evolution of the EPR spectra of sample 5. | ||
| Sample number | Polaron areaa | NiO/Polaron area ratio | χ m T b /emu K mol−1 | H c/Gc | ||
|---|---|---|---|---|---|---|
| 4.2 K | 300 K | 4.2 K | 300 K | |||
| a Area of the EPR signal corresponding to the organic polaron. The numbers in brackets are values normalized to that of sample 5. b Measured at 300 K and 100 G. c Measured at 2 K. d Data at 50 K. | ||||||
| 1 | 0.376 (2) | 0.037 (7) | 5 | 117 | 140 | 510 |
| 2 | 0.795 (4) | 0.049 (9) | 4 | 124 | 14 | 550 |
| 3 | 0.000 (0) | 0.000 (0) | — | — | 40 | 450 |
| 4 | 0.000 (0) | 0.000 (0) | — | — | 40 | 450 |
| 5 | 18.317 (100) | 0.517 (100) | 1 | 50 | 0.115 | 500 |
| 6 | 0.797 (4) | 0.163d (32) | 86 | 409 | 0.409 | 435 |
| 7 | 4.309 (24) | 0.161 (31) | 21 | 859 | 0.285 | 380 |
Superimposed on this background, we occasionally observed a sharp EPR peak (3360 G, ΔH∼20 G) with considerably less area at r.t. Ni2+-exchanged zeolite or aluminosilicate submitted to analogous thermal treatment do not exhibit any narrow EPR peak. FT-Raman spectroscopy also indicates that the samples are free from the presence of detectable monomer. Thus, based on the spectroscopic characterisation of the samples that indicates polyalkyne formation and on the g value, we attribute the narrow signal (g=2.0038 for sample 1 measured at 4.2 K) to the presence of polarons generated by spontaneous polyacetylene or polypropyne doping, concurrent with the alkyne polymerisation process. Typically, the intensity of this sharp signal increases with the polymerisation temperature and the carbon content of the sample, but the dependence is not linear. Table
2 also lists the relative area of the polaron signal measured for each sample. There is again no relationship between the polaron population and the reaction temperature. This is compatible with the existence of a threshold below which no polarons are formed. In contrast to the background signal attributed to NiO, the intensity of the sharp peak increases linearly with 1/T, following the Curie law, as expected for isolated polarons having a paramagnetic coupling (Fig. 4).
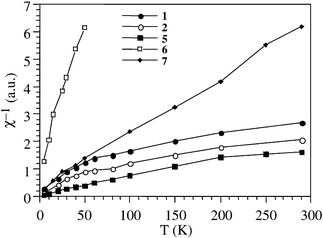 | ||
| Fig. 4 Thermal variation of the inverse of the dynamic susceptibility (χ), measured from the area of the narrow signal, showing their paramagnetic behaviour for the mordenite samples. Samples 3 and 4 do not exhibit a measurable polaron signal. | ||
SQUID measurements of the samples are dominated by the conducting behaviour of the antiferromagnetic species (Fig. 5). The metallic behaviour of most of the samples can be attributed to the presence of NiO aggregates. The contribution of polyacetylene polarons to the bulk magnetic susceptibility of the samples is very small compared to that of the NiO clusters and thus can be ignored. The magnetic susceptibility decreases with the recording temperature, in agreement with the observed behaviour in the EPR. A residual magnetization at zero field Hc was also observed (see Table 2) and the NiO- containing samples exhibit hysteresis.
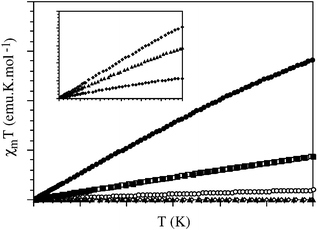 | ||
| Fig. 5 Thermal variation of the product of the magnetic susceptibility and the temperature for all the mordenite samples. (•), (○), (■), (□), (✦), (◊) and (▲) correspond to samples 1 to 7, respectively. | ||
Besides mordenite, the possibility of spontaneous doping concomitant with the acetylene polymerisation process was also studied in Ni2+-exchanged Al/MCM-41. The characteristics of these samples are also included in Table 1, the most relevant parameter being the remarkably high polymer loading achieved for samples 9 and 10 and the absence of polymer in sample 11. The ability of zeolites to generate radical cations as persistent species has been ascribed on the one hand to the presence of acid sites12,17 and on the other to the tight fit of the zeolite framework that protects the species from the attack of external nucleophiles.18 According to this widely-assumed rationalisation, Al/MCM-41, having only weak acid sites and large mesopores, should be a much less suitable support for the observation of persistent organic radical ions than conventional microporous zeolites. Thus, it is not surprising that in certain cases Al/MCM-41 has been reported to be inactive for the generation of radical ions.12 However, we have measured for our Al/MCM-41 samples containing polyacetylene, the presence of a remarkably high polaron population, comparable to that of mordenite samples 1 and 2 (Fig. 6). Relative to sample 5 of mordenite, the areas of the organic polaron at room temperature were 0, 4 and 5% for samples 8, 9 and 10, respectively.
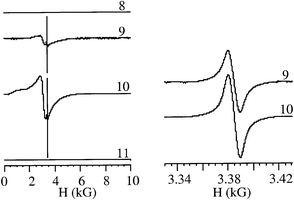 | ||
| Fig. 6 Left: EPR spectra of the MCM-41 samples at room temperature. The areas of the polaron signal corresponding to samples 9 and 10 were 4 and 5% that of sample 5 recorded under the same conditions. Right: An expansion of the narrow signals corresponding to organic polarons. | ||
A likely explanation, which would reconcile our observation with the previous reports, is that in our case essentially all the pore volume of the Al/MCM-41 channels is filled with polymer and, therefore, polarons are protected from oxygen and ambient moisture. To provide an idea of the persistence of the polarons in our Al/MCM-41 samples, we repeated the EPR and SQUID measurements on samples stored in vials under air for 3 months and found no noticeable variation in the intensity of the signals.
To determine if it is possible to dope polyacetylene chemically to a larger extent than spontaneous doping concurrent with polymer formation, polymerisation was carried out at three different reaction temperatures with the previously hydrogen-reduced, Ni(0),H-mordenite. Two important differences using Ni(0) mordenite compared to Ni2+-mordenite were observed. Firstly, the magnetic susceptibility was reduced by 2–3 orders of magnitude, the response of the powder being characteristic of a paramagnetic solid. Thus, the χT value of samples 5, 6 and 7 was 0.1–0.4, much closer to the expected value for a S=1/2 spin state.
Secondly, the intensity of the sharp peak corresponding to polyacetylene polarons for sample 5 was remarkably high. Thus, taking into account that purely Brønsted H-mordenite only forms lesser amounts of polyacetylene, we attribute the higher polyacetylene loadings to the presence of nickel active sites and the increase in the EPR polaron signal to the chemical doping according to eqn. (2). Although both p- and n- polarons show exactly the same EPR signal and cannot be distinguished by SQUID measurements; the charge of the polaron, at least for the Ni(0) mordenite sample, must be negative, corresponding to n-doping.
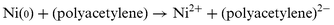 | (2) |
However, the fact that the density of the n-polarons is much below the maximum stoichiometric amount according to the Ni content does not allow us to rule out that for Ni2+-mordenite the sign of the polaron charge is positive. It has to be noted, however, that it is generally assumed that zeolite and porous aluminosilicates stabilise positively charged species and that negative species cannot be present within the internal voids.23 In addition, radical cations and not radical anions are the species observed in zeolites. However, these two general facts are related to the Al content of the zeolite framework and to the density of framework negative charges. In our case, it has to be noted that the Al content of our samples is much lower than that of faujasites X (one Al per Si) or Y (one Al per 2.4 Si). Although not common, the incorporation of negatively charged species, in zeolites having a low Al content has been reported.24 In this case hydroxide anions located inside the channels of modified Al/MCM-41 were found to be effective as a heterogeneous basic catalyst.
In conclusion, we have shown that alkyne in situ polymerisation is accompanied by the formation of NiO clusters and partial doping of the polymer. Polarons are persistent over extended periods of time. The extent of the doping increases with polymerisation temperature, but is not linear with respect to the carbon content of the samples. Spontaneous doping of polyacetylene also occurs in Al/MCM-41. Finally, we have found that Ni(0) clusters can act concurrently as polymerisation sites and as reducing agents to produce n-polarons to a significant extent.
Experimental
Mordenite with a Si/Al ratio of 12 was a commercial sample (P.Q. Industries CBV 20A). MCM-41 with a Si/Al ratio of 13 was synthesised by hydrothermal crystallisation from aerosil and alumina using hexadecyltrimethylammonium bromide as structure directing agent and tetramethylammonium hydroxide as mineraliser as previously reported.25–27 The structure of MCM-41 was confirmed by powder X-ray diffraction and the BET surface area (840 m2 g−1) and micropore volume (0.4 ml g−1) were measured by isothermal N2 and Ar adsorption of the sample.Ni2+-ion exchange was accomplished by stirring magnetically at room temperature for 2 h a suspension of Ni(OAc)2 in water (177 g×l−1) in the presence of the aluminosilicate, using a liquid/solid ratio of 10. The solid was filtered and washed thoroughly with distilled water. The pH of the Ni(OAc)2 solution was controlled by adding acetic acid. For samples 1 and 5 the pH was not controlled.
Polymerisation was carried out by contacting the monomer with dehydrated zeolite at the appropriate temperature for 1 h. Prior to the polymerisation, the corresponding zeolite (250 mg) was dehydrated by heating at 300°C under 1 Torr pressure for 1 h. The zeolite was then equilibrated at the required temperature with a thermostatted silicone bath and acetylene or propyne was admitted into the flask at atmospheric pressure. The temperature of the reaction was measured by a thermocouple in contact with the powder. Commercial acetylene was purified by passing consecutively through an aqueous solution of 60% sulfuric acid and through solid NaOH beads.
Combustion C and H analyses of the samples were carried out in a Fisons EA-1108 elemental analyser. Diffuse reflectance UV–Vis spectra were recorded in a Cary 5G spectrophotometer adapted with a praying mantis using BaSO4 as standard. FT Raman spectra were obtained with a Bio-Rad Raman II spectrophotometer. The 1.064 mm line of a Nd:YAG laser was used for excitation and a Ge detector was used. EPR spectra were recorded on a Bruker ER200D spectrometer working at X band (9.65 GHz) using DPPH (g=2.0036) as reference. To quantify the relative intensity of the EPR signals weighed amounts of the powders (∼10 mg) were placed in the cell. The area of the spectrum was obtained by double integration of the signal and corrected for the corresponding weight of sample. The magnetic susceptibility was measured using a Quantum Design MPMS-XL-5 susceptometer equipped with a SQUID sensor.
Acknowledgements
Financial support by the European Union (Brite Euram III, project CHARMED) is gratefully acknowledged. The authors thank Profs. A. Gilbert and D. Cardin for their collaboration in this Brite project and for disclosing part of their electron microscopy study. HG also thanks the Spanish DGICYT for funding through an operating grant.References
- T. Bein, Stud. Surf. Sci. Catal., 1996, 102, 295–321 Search PubMed.
- E. Ruiz-Hitzky and P. Aranda, An. Quim., Int. Ed., 1997, 93, 197–212 Search PubMed.
- J. C. W. Chien, Polyacetylene: Chemistry Physics and Materials Science, Academic Press, New York, 1984. Search PubMed.
- S. D. Cox and G. D. Stucky, J. Phys. Chem., 1991, 95, 710 CrossRef CAS.
- C. Pereira, G. T. Kokotailo and R. J. Gorte, J. Phys. Chem., 1991, 95, 705–09 CrossRef CAS.
- G. J. Millar, A. R. Lewis, G. A. Bowmaker and R. P. Cooney, J. Mater. Chem., 1993, 3, 867–72 RSC.
- A. R. Lewis, G. J. Millar, R. P. Cooney and G. A. Bowmaker, Chem. Mater., 1993, 5, 1509–17 CrossRef CAS.
- D. Cardin, S. Constantine, A. Gilbert, A. Lay, M. S. Galletero, M. Alvaro, H. García and F. Márquez, J. Am. Chem. Soc., 2001, 123, 3141–42 CrossRef CAS.
- F. R. Chen and J. J. Fripiat, J. Phys. Chem., 1992, 96, 819–23 CrossRef CAS.
- V. Martí, V. Fornés, H. García and H. D. Roth, Eur. J. Org. Chem., 2000, 3, 473–77 CrossRef.
- H. García, V. Martí, I. Casades, V. Fornés and H. Roth, Phys. Chem. Chem. Phys., 2001, 13, 2955–60 RSC.
- A. Corma, V. Fornés, H. García, V. Martí and M. A. Miranda, Chem. Mater., 1995, 7, 2136–43 CrossRef CAS.
- A. Corma and H. García, Top. Catal., 1998, 6, 127–40 Search PubMed.
- J.-V. Folgado, H. García, V. Martí and M. Esplá, Tetrahedron, 1997, 53, 4947–56 CrossRef CAS.
- V. Martí, L. Fernández, V. Fornés, H. García and H. D. Roth, J. Chem. Soc., Perkin Trans. 2, 1999, 2, 145–52 RSC.
- J. V. Caspar, V. Ramamurthy and D. R. Corbin, J. Am. Chem. Soc., 1991, 113, 600–10 CrossRef CAS.
- X. Liu, K.-K. Iu, J. K. Thomas, H. He and J. Klinowski, J. Am. Chem. Soc., 1994, 116, 11811–18 CAS.
- V. Ramamurthy, J. V. Caspar and D. R. Corbin, J. Am. Chem. Soc., 1991, 113, 594–600 CrossRef CAS.
- V. Ramamurthy, D. F. Eaton and J. V. Caspar, Acc. Chem. Res., 1992, 25, 299–306 CrossRef CAS.
- V. Ramamurthy, Chimia, 1992, 46, 359–76 Search PubMed.
- E. A. Piocos, D. W. Werst, A. D. Trifunac and L. A. Eriksson, J. Phys. Chem., 1996, 100, 8408–17 CrossRef CAS.
- D. MacInnes, M. A. Druy, P. J. Nigrey, D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, Chem. Commun., 1981, 317–19 RSC.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- I. Rodriguez, S. Iborra, A. Corma, F. Rey and J. L. Jordà, Chem. Commun., 1999, 7, 593–94 RSC.
- P. Behrens and M. Haak, Angew. Chem., Int. Ed. Engl., 1993, 32, 696–99 CrossRef.
- P. Behrens, Adv. Mater., 1993, 5, 127–32 CrossRef CAS.
- D. W. Breck, Zeolite Molecular Sieves: Structure, Chemistry and Use, Wiley, New York, 1974. Search PubMed.
| This journal is © the Owner Societies 2002 |