Cryptophanols, new versatile compounds for the synthesis of functionalized cryptophanes and polycryptophanes†
Magali
Darzac
a,
Thierry
Brotin
*a,
Denis
Bouchu
b and
Jean-Pierre
Dutasta
*a
aStéréochimie et Interactions Moléculaires, École Normale Supérieure de Lyon (UMR CNRS 5532), 46 Allée d′Italie, F-69364, Lyon Cedex 07, France.. E-mail: dutasta@ens-lyon.fr;; Fax: 33(0)472728483
bCentre de Spectrométrie de Masse, Université Claude Bernard Lyon I, 43 Boulevard du 11 Novembre 1918, F-69622, Villeurbanne Cedex, France
First published on 14th December 2001
Abstract
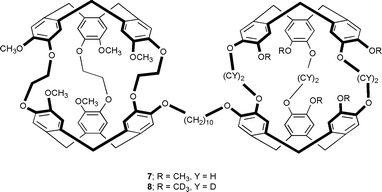
After deprotection with a palladium catalyst, mono-allylated cryptophane-A (1, 2) and cryptophane-E (3) gave the new cryptophanols 4, 5 and 6, respectively, which are important key compounds for the preparation of mono-functionalized cryptophanes as well as for the design of large supramolecular hosts such as the bis-cryptophanes 7 and 8.‡
Cryptophanes are fascinating molecules and represent an important class of supramolecular hosts known to form stable inclusion complexes with neutral and cationic molecules.1,2 This was recently enlightened with the exceptionally large affinity of cryptophane-A for xenon in tetrachloroethane solution (K ≈3000 M−1 at 293 K).3 They are therefore of potential interest for the development of new chemical sensors in the field of molecular recognition and especially for applications of xenon NMR in biological systems.4 This requires however the design of complex molecular receptors using cryptophanes as the host. So far, little effort has been made to develop the chemistry of cryptophanes exhibiting specific functionality and bio-compatibility, or to build large supramolecular systems with e.g. polymeric or dendritic structures. The present work reports the synthesis of mono-functionalized cryptophanes 1–3 bearing a single allyl group, which are easily converted to the mono-hydroxylated compounds 4–6. Hosts 4–6 are very attractive and important key compounds for the synthesis of original substituted cryptophanes, such as chiral precursors and bio-compatible ligands for molecular recognition, as well as for the generation of large supramolecular systems. As an example we report the synthesis of symmetrical (7) and unsymmetrical (8) bis-cryptophanes, which have been fully characterized by usual analytical techniques.
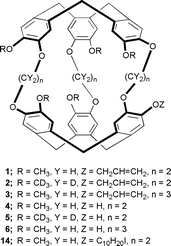
The substitution of only one protecting group in place of a methoxy peripheral substituent in the cryptophane molecules requires some restrictions. Firstly, this group has to be very stable under acidic condition as the last step of the synthesis is usually performed in formic acid, and secondly, it has to be easily cleaved. In addition this group has to be of small size as previous work has shown that bulky substituents attached to the cryptophane precursors, such as long alkyl chains, prevent the formation of the cryptophanes in the final cyclization step. This results in a rather limited number of available efficient protecting groups, even though the protection of phenol has been widely described in the literature.5
The choice of an adequate protecting group was thus suggested and facilitated by our recent investigations on the formation of cryptophanes from their precursors by mass spectrometry. The behaviour of cryptophane precursors under the LSIMS conditions showed some similarities with chemistry in solution, and it was demonstrated that both the allyl and benzyl moieties could be used as potential protecting groups without altering the last cyclization step.6 In addition these groups are easily cleaved by various methods.
The attachment of a single substituent requires a modification of the strategy used for cryptophanes with C3 or D3 symmetry. For this purpose we followed our procedure recently described for the preparation of deuterium labelled cryptophanes.7 This method based on a multistep synthesis involves the formation of the two cyclotriveratrylene units at two different stages of the preparation as depicted for 4 in Scheme 1.
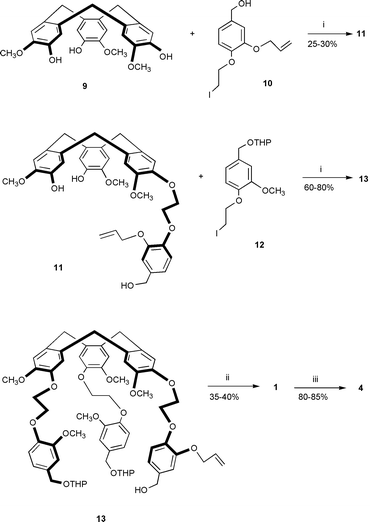 | ||
| Scheme 1 Reagents and conditions: i, Cs2CO3, DMF, 80 °C, 18 h; ii, CHCl3, HCO2H (50∶50), 55 °C, 2 h 30 min; iii, Pd(OAc)2, PPh3, THF, NHEt2, H2O, 4 h. | ||
Cyclotriveratrylene 9 was prepared according to a known procedure8 and was allowed to react with the mono-protected benzyl alcohol 10 in the presence of caesium carbonate as a base to give compound 11 in 25–30% yield. This low yield was mainly due to the formation of the di- and tri-substituted derivatives. Reaction of 11 with 12 under similar conditions gave the precursor 13 in good yields (60–80%). The use of the tetrahydropyranyl (THP) ether function made the purification easier of the desired material by column chromatography on silica gel. A similar strategy was applied for the preparation of cryptophanes 2 and 3 to give the corresponding precursors with yields ranging from 60 to 80%.
The key step in this synthesis is the trimerization of the benzyl alcohol groups of the precursors to give cryptophanes 1–3. Attempts to use classical reagents such as perchloric acid in methanol or pure formic acid were unsuccessful and led to non-isolable compounds. However, a mixture of chloroform and formic acid as solvent (50∶50) at 55 °C was found to be effective for leaving the protecting group unreacted and led to the formation of the desired compounds 1–3 in 35–40% yields.§ These yields are however significantly lower than that obtained with cryptophane-A under the same experimental conditions.2
The difficulties encountered in the preparation of these new derivatives led us to the conclusion that new routes using milder conditions should also be examined. For instance it has been reported that scandium triflate promotes calixarene formation in moderate yields under mild conditions leaving unreacted allyl groups.9 In the light of these results, attempts were made to reproduce these experiments with cryptophane precursors. The results appeared promising as, for instance, reaction of 13 with scandium triflate (0.6 equiv.) in acetonitrile gave 1 in 10% yield after purification. Finally, removal of the allyl group was achieved by the use of a palladium catalyst in the presence of diethylamine and water to give the unprotected cryptophanes 4–6 in 80–85% yields.§ These new cryptophanes have been isolated as their anti chiral isomer and are of large potential interest in the design of new supramolecular hosts.
Hosts 7 and 8 were designed for the study of the guest exchange processes between cryptophanes and exemplify the potentialities of cryptophanols 4–6. Compounds 7 and 8 are similar except that one cryptophane in molecule 8 has been replaced by its partially deuterated congener. Both compounds have been obtained by a Williamson synthesis using caesium carbonate as base and 1,10-diiododecane as alkylating agent. Compound 7 was obtained in 30% yield in DMF from two equivalents of 4 and one equivalent of 1,10-diiododecane. The unsymmetrical molecule 8 required the synthesis of compound 14, which was obtained by treating 4 with an excess of 1,10-diiododecane. The reaction of 14 and cryptophanol 5 under similar conditions, afforded compound 8.§ Bis-cryptophanes 7 and 8 have been isolated as a mixture of stereoisomers. 1H, 13C NMR and MALDI mass spectrometry were used to ascertain the structures of molecules 7 and 8 (Fig. 1).
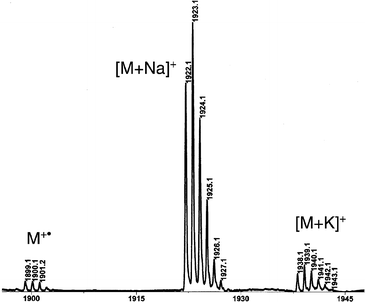 | ||
| Fig. 1 MALDI spectrum of 8. | ||
In summary, we report here the preparation of the new cryptophanols 4–6 containing a single hydroxy function. The strategy described herein provides the basis for a general method for preparing functionalized cryptophanes. These new molecules are believed to be key compounds for the elaboration of large supramolecular systems, as suggested by the synthesis of compounds 7, 8 and 14. Monofunctionalized cryptophanes provide an original tool for the optical resolution of new diastereoisomeric derivatives via the reaction of molecules 4–6 with a chiral substrate.10 We now focus our work on the synthesis of selectively multiprotected cryptophanes for the design of new elaborate structures such as hosts of biological relevant interest, water-soluble cryptophanes and xenon@cryptophane complexes.
We thank J.-C. Mulatier for skilful experimental assistance.
Notes and references
- L. Garel, B. Lozach, J.-P. Dutasta and A. Collet, J. Am. Chem. Soc., 1993, 115, 11652 CrossRef CAS; L. Garel, J.-P. Dutasta and A. Collet, Angew. Chem., Int. Ed. Engl., 1993, 32, 1169 CrossRef; A. Collet, in Comprehensive Supramolecular Chemistry,Pergamon, New York, 1996, vol. 2, p. 325. Search PubMed.
- A. Collet, J.-P. Dutasta, B. Lozach and J. Canceill, Topics in Current Chemistry, 1993, 165, 103 Search PubMed; A. Collet, J.-P. Dutatsa and B. Lozach, Bull. Soc. Chim. Belg., 1990, 99, 617 Search PubMed.
- K. Bartik, M. Luhmer, J.-P. Dutasta, A. Collet and J. Reisse, J. Am. Chem. Soc., 1998, 120, 784 CrossRef CAS.
- C. Dybowski and N. Bansal, Annu. Rev. Phys. Chem., 1991, 42, 433 CrossRef CAS; P. J. Barrie, J. Klinowski and D. Raftery, Prog. Nucl. Magn. Reson. Spectros., 1992, 91 Search PubMed; M. Luhmer, B. M. Goodson, Y. Q. Song, D. D. Laws, L. Kaiser, M. C. Cyrier and A. Pines, J. Am. Chem. Soc., 1998, 120, 3502.
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 2nd edn., 1991. Search PubMed.
- T. Brotin, M. Darzac, D. Forest, M. Becchi and J.-P. Dutasta, J. Mass Spectrom., 2001, 36, 1092 CrossRef CAS.
- T. Brotin, A. Lesage, L. Emsley and A. Collet, J. Am. Chem. Soc., 2000, 122, 1171 CrossRef CAS; T. Brotin, T. Devic, A. Lesage, L. Emsley and A. Collet, Chem. Eur. J., 2001, 7, 1561 CrossRef CAS.
- J. Canceill, A. Collet and G. Gottarelli, J. Am. Chem. Soc., 1984, 106, 5997 CrossRef CAS.
- H. Konishi, H. Sakakibara, K. Kobayashi and O. Morikawa, J. Chem. Soc., Perkin Trans. 1, 1999, 2583 RSC.
- T. Brotin, M. Darzac, R. Barbe and J.-P. Dutasta, unpublished results..
Footnotes |
| † Electronic supplementary information (ESI) available: experimental details for compounds 4, 5, 6, 7, 8 and 14. See http://www.rsc.org/suppdata/cc/b1/b109301k/ |
| ‡ Presented at the 6th International Conference on Calixarenes, May 29–June 2, 2001, Twente (NL). |
| § The cryptophanes described herein were purified by column chromatography on silica gel and recrystallized in a chloroform–ethanol mixture. All new compounds were fully characterized by 1H and 13C NMR spectroscopy and by HRMS. The detailed synthesis of 4–8 and 14 will be presented in a forthcoming full paper. |
| This journal is © The Royal Society of Chemistry 2002 |