Palladium-catalysed asymmetric arylation of tert-cyclobutanols via enantioselective C–C bond cleavage
Takahiro
Nishimura
,
Satoshi
Matsumura
,
Yasunari
Maeda
and
Sakae
Uemura
*
Department of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Sakyo-ku, Kyoto, 606-8501, Japan. E-mail: uemura@scl.kyoto-u.ac.jp; Fax: 81 75 753 3573; Tel: 81 75 753 5687
First published on 4th December 2001
Abstract
Palladium-catalysed arylation of tert-cyclobutanols with aryl bromide involving enantioselective C–C bond cleavage affords chiral ketones with moderate to good enantioselectivity.
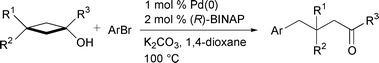
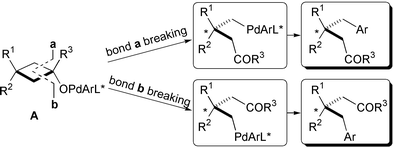
Transition metal-catalysed asymmetric reactions have been used as modern and powerful methods for the synthesis of optically active compounds, mainly by a carbon–hydrogen, a carbon–heteroatom, and a carbon–carbon bond formation to create a new chiral centre on carbon.1 In sharp contrast, although a metal-catalysed selective C–C bond cleavage reaction has been found in recent years,2 the example of the enantioselective C–C bond cleavage reaction has been scarcely reported to the best of our knowledge.3 We have recently disclosed a Pd(0)-catalysed arylation of tert-cyclobutanols involving β-carbon elimination from an intermediate arylpalladium(II)-alcoholate to afford arylated ketones [eqn. (1)].4 This ring
 | (1) |
 | ||
| Scheme 1 | ||
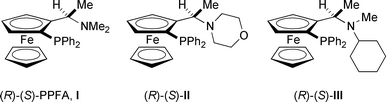
In our preliminary communication of this novel reaction, the produced ketones were racemic in spite of the use of (R)-(+)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) as a chiral phosphine ligand. In the search for a successful asymmetric arylation involving an enantioselective C–C bond cleavage using several chiral phosphine ligands, we found that some monophosphine ligands work effectively to give the chiral ketones in high yields with moderate to good enantiomeric excess. We wish to report here the effective asymmetric arylation of 3-substituted tert-cyclobutanols.
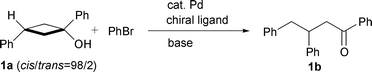
First, the arylation of tert-cyclobutanol 1a was carried out using some commercially available chiral bisphosphine ligands, such as (R)-Tol-BINAP, (R)-(S)-BPPFA, (+)-Me-DUPHOS and (+)-DIOP, in place of BINAP. Only slight stereoselectivity was observed (∼2% ee), but the product yields were quite low [eqn. (2)]. Interestingly, monophosphine ligands such
 | (2) |
| Entry | Ligand | PhX | Solvent (mL) | Temp. (°C) | Time (h) | GLC yield (%)b | ee (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1a (0.10 mmol), Pd(OAc)2 (0.005 mmol), ligand (0.02 mmol), PhX (0.12 mmol), Cs2CO3 (0.12 mmol), under N2. 1a was completely consumed in entries 8 and 9. b Based on 1a employed. c Isolated yield. d K2CO3 was used as a base. | |||||||
| 1 | I | PhOTf | THF (1.0) | 90 | 24 | 42 | 59 |
| 2 | I | PhOTf | 1,4-dioxane (1.0) | 90 | 24 | 12 | 52 |
| 3 | I | PhOTf | toluene (1.0) | 90 | 24 | 70 | 48 |
| 4 | I | PhOTf | toluene (0.5) | 70 | 72 | 24 | 57 |
| 5 | I | PhBr | toluene (0.5) | 70 | 72 | 71c | 59 |
| 6 | I | PhBr | toluene (0.5) | 80 | 48 | 70c | 58 |
| 7d | I | PhBr | toluene (0.5) | 80 | 48 | 11 | 57 |
| 8 | II | PhBr | toluene (0.5) | 80 | 48 | 98c | 49 |
| 9 | III | PhBr | toluene (0.5) | 80 | 48 | 93c | 74 |
The results of the asymmetric arylation of several monocyclic tert-cyclobutanols leading to chiral γ-arylated ketones under the optimised conditions described above are listed in Table 2.† Using bromobenzene as arylating agent under the conditions composed of Pd(OAc)2, ligand III,‡ and Cs2CO3 in toluene, the substrate alcohols were completely consumed within 24 h, and 3-substituted cyclobutanols 1a, 2a, and 3a gave the corresponding γ-arylated ketones 1b, 2b, and 3b in high yields with moderate to good enantioselectivity, respectively (entries 1, 5 and 6). The lower reaction temperature (60 °C) did not affect the ee value (entry 2). The arylation of 1a with p-bromochlorobenzene or p-bromotoluene also occurred smoothly to give 1c and 1d in high yields with good ee value (entries 3 and 4). Similarly, the reaction of 3a with 2-bromonaphthalene afforded 3c in high yield (entry 7). It should be noted that this arylation could also be applied to the 1-alkyl-substituted cyclobutanol 4a, and the corresponding dialkylketone 4b was obtained in high yield with moderate enantioselectivity (entry 8). 3-Disubstituted cyclobutanols gave the corresponding ketones having quaternary carbon centres.7 Treatment of cyclobutanol 5a8 under the same conditions for 47 h afforded (−)-69 with 61% ee, while the isomer 5b8 gave (+)-69 with 43% ee when reacted for 37 h [eqns. (3) and (4)]. These results suggest
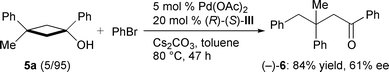 | (3) |
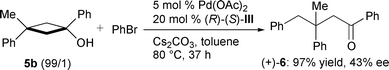 | (4) |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate (cis/trans) | Ar | Product and isolated yield (%) | ee (%)b | |
| a Reaction conditions: alcohol (0.2 mmol), Pd(OAc)2 (0.01 mmol), (R)-(S)-III (0.04 mmol), aryl bromide (0.24 mmol), Cs2CO3 (0.24 mmol), toluene (1 mL), 80 °C, 24 h under N2. b Determined by HPLC. c 60 °C, 48 h. d 42 h. | |||||
| 1 |

|
Ph |
|
99 | 77 |
| 2c | 1a (98/2) | Ph | 1b | 83 | 78 |
| 3 | p-ClC6H4 | 1c | 90 | 73 | |
| 4 | p-MeC6H4 | 1d | 91 | 75 | |
| 5 |
 2a (96/4)
2a (96/4) |
Ph |
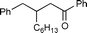 2b
2b
|
99 | 64 |
| 6 |
 3a (97/3)
3a (97/3) |
Ph |
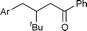 3b
3b
|
99 | 53 |
| 7 | 2-naphthyl | 3c | 92 | 60 | |
| 8d |
 4a (99/1)
4a (99/1) |
Ph |
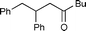 4b
4b
|
86 | 60 |

In summary, we have described the novel asymmetric arylation of tert-cyclobutanols involving C–C bond cleavage, in which the possibility of enantioselective C–C bond cleavage was demonstrated using some chiral ligands. Our study for finding a more efficient stereocontrolled catalytic system for this reaction is now in progress.
Notes and references
- I. Ojima, Catalytic Asymmetric Synthesis, 2nd ed., Wiley-VCH, New York, 2000 Search PubMed.
- M. Murakami and Y. Ito, in Activation of Unreactive Bonds and Organic Synthesis, ed. S. Murai, Springer, New York, 1999, pp. 97–129 Search PubMed.
- Rhodium-catalysed asymmetric hydrogenolysis of the α-carbon–carbon bond of cyclobutanone involving an enantioselective C–C bond cleavage was presented by M. Murakami, H. Amii, and Y. Ito, at the 69th Annual Meeting of the Chemical Society of Japan, March 1995, Kyoto, Abstract II p.1127.
- T. Nishimura and S. Uemura, J. Am. Chem. Soc., 1999, 121, 11010 CrossRef CAS.
- (a) Y. Uozumi and T. Hayashi, J. Am. Chem. Soc., 1991, 113, 9887 CrossRef CAS; (b) Y. Uozumi, N. Suzuki, A. Ogiwara and T. Hayashi, Tetrahedron, 1994, 50, 4293 CrossRef CAS.
- T. Hayashi, T. Mise, M. Fukushima, M. Kagotani, N. Nagashima, Y. Hamada, A. Matsumoto, S. Kawakami, M. Konishi, K. Yamamoto and M. Kumada, Bull. Chem. Soc. Jpn., 1980, 53, 1138 CAS.
- E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388 CrossRef.
- Alcohols 5a and 5b were separated by high performance preparative liquid chromatography. However, the exact stereochemistry (cis or trans) is not yet clear. The stereochemistry presented here was presumed from the 1H-NMR of an analogous alcohol.
- Specific rotation of 6; (−)-6: [α]25D = −10.2 (c = 0.5, in CHCl3); (+)-6; [α]25D = +4.2 (c = 0.5, in CHCl3).
Footnotes |
| † Experimental: A mixture of Pd(OAc)2 (0.01 mmol), (R)-(S)-III (0.04 mmol), Cs2CO3 (0.24 mmol) and toluene (0.5 mL) in a 10 mL two-necked flask was stirred at rt under N2. After 0.5 h, aryl bromide (0.24 mmol) and alcohol (0.20 mmol) in toluene (0.5 mL) were added and the mixture was stirred at 80 °C until the reaction had reached completion by monitoring with TLC analysis. The reaction mixture was cooled to rt and then filtered through a pad of Florisil. The filtrate was concentrated under vacuum to give an oil, which was subjected to column chromatography on SiO2 with EtOAc–hexane (2 ∶ 98) as eluent. The enantiomeric excess was determined by HPLC using Daicel Chiralcel® AD and OD columns (4.6 × 250 mm, 3% propan-2-ol–hexane) at 25 °C. |
| ‡ Analytical data for ligand III: Ligand III is a new compound synthesised from (R)-(S)-PPFOAc and N-methylcyclohexylamine according to the reported procedure, ref 6. Yellow solid; mp 159.2–160.0 °C; [α]25D = −286.9 (c = 0.5, CHCl3); IR (KBr) 2956, 2939, 2774, 1446, 1433, 818, 749, 739, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.49–1.58 (m, 10H), 1.29 (d, J = 6.3 Hz, 3H), 1.65 (s, 3H), 2.15–2.30 (m, 1H), 3.70 (s, 1H), 3.73–4.26 (m, 4H), 3.94 (s, 5H), 7.01–7.70 (m, 10H). 31P NMR (161.9 MHz, CDCl3) δ 31.3. Anal. Calcd. for C31H36FeNP: C, 73.09; H, 7.12; N, 2.75; Found: C, 73.02; H, 7.07; N, 2.82%. |
| This journal is © The Royal Society of Chemistry 2002 |