Functionalisation of carbon dioxide by an iron(II) complex†
Leslie D.
Field
*,
Warren J.
Shaw
and
Peter
Turner
School of Chemistry, University of Sydney, New South Wales, 2006, Australia.. E-mail: l.field@chem.usyd.edu.au.
First published on 14th December 2001
Abstract
Addition of carbon dioxide to trans-Fe(dmpe)2(SCHNEt)H 2 affords the iminium carboxylate trans-Fe(dmpe)2(SCHN+(Et)CO2−)H 4, which rearranges to the ferracyle cis-Fe(dmpe)2(SCH2N(Et)C(O)O-κS,O) 5.
The fixation and functionalisation of carbon dioxide at transition metal centres has received much recent attention due to the potential of this greenhouse gas as an abundant and inexpensive source of carbon in the construction of more complex organic compounds.1
We recently reported the insertion reaction between the iron hydride cis-Fe(dmpe)2H21 (dmpe = Me2PCH2CH2PMe2) and the heteroallenes carbon dioxide (CO2), carbon disulfide (CS2), carbonyl sulfide (COS),2 isocyanates (RNCO) and isothiocyanates (RNCS)3 to form η1-bound (thio)formato and (thio)formimidato-type ligands. Insertion into both iron–hydride bonds of 1 occurred in the presence of excess heteroallene in each case except for that of ethyl or methyl isothiocyanate, where a N-to-C condensation of the heteroallene resulted from nucleophilic attack by the coordinated N-alkylthioformimidate of trans-Fe(dmpe)2(SCHNR)H (R = Me, Et (2)) on another RNCS molecule to give the free zwitterion trans-Fe(dmpe)2(SCHN+(R)C(S)N−(R))H (R = Me, Et (3)) (Scheme 1).3
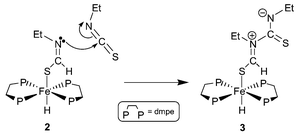 | ||
| Scheme 1 Dimerization of EtNCS at an iron(II) centre.3 | ||
In this communication, we report initial studies on the reaction between CO2 and trans-Fe(dmpe)2(SCHNEt)H 2 to form an intermediate iron(II) iminium carboxylate zwitterion which rearranges, by insertion/cyclisation at the iron(II) centre, to form a novel heteroatomic ferracycle (Scheme 2).
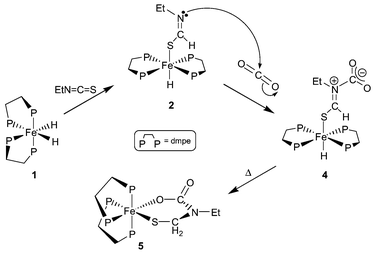 | ||
| Scheme 2 Reaction between 1 and EtNCS and CO2 sequentially. | ||
Addition of CO2 (1 atm) to a degassed solution of trans-Fe(dmpe)2(SCHNEt)H 2, formed in situ by the insertion reaction between cis-Fe(dmpe)2H21 and EtNCS (Scheme 2), in toluene-d8 at 300 K afforded the zwitterionic iminium carboxylate complex, trans-Fe(dmpe)2(SCHN+(Et)CO2−)H 4 as the major kinetic product (Scheme 2). The 31P{1H} NMR spectrum of trans-Fe(dmpe)2(SCHN+(Et)CO2−)H 4 comprised a singlet resonance (δ 69.4) for the equivalent phosphorus donors. In the 1H NMR spectrum, the hydride resonance (δ −25.20) appeared as a pentet (2JPH 50 Hz), while the proton of the iminium fragment occurred as a singlet at δ 9.30. The iminium and carboxylate 13C nuclei resonated at δ 172.5 (unresolved 31P coupled multiplet) and 174.6, respectively.
An X-ray diffraction study of a red prismatic single crystal, grown from a concentrated toluene-d8 solution at 300 K, confirmed the zwitterionic nature of 4 (Fig. 1).‡ The dmpe ligands adopted at least two complementary orientations, the populations of which were refined and fixed. The S(1)–C(1) bond length (1.670(2) Å) is extremely short for a C–S single bond (typically ∼1.8 Å4). The C(1)–N(1), N(1)–C(2) and C–O bond lengths also suggest delocalisation of the zwitterionic charges throughout the dimer ligand.
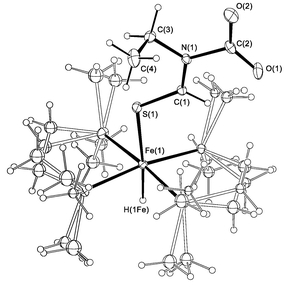 | ||
| Fig. 1 ORTEP5 diagram of 4 showing 20% displacement ellipsoids and disorder in the dmpe ligands. Selected bond distances (Å) and angles (°): Fe(1)–S(1) 2.2925(10), S(1)–C(1) 1.670(2), C(1)–N(1) 1.321(3), N(1)–C(2) 1.518(3), C(2)–O(1) 1.232(3), C(2)–O(2) 1.226(3); Fe(1)–S(1)–C(1) 115.76(8), S(1)–C(1)–N(1) 127.16(17), N(1)–C(2)–O(1) 113.8(2), N(1)–C(2)–O(2) 113.3(2). | ||
In solution at 300 K, trans-Fe(dmpe)2(SCHN+(Et)CO2−)H 4 quantitatively isomerised to a new compound 5 with a cis disposition of the dmpe ligands, and with no metal hydride (Scheme 2). Compound 5 was identified as the ferracycle cis-Fe(dmpe)2(SCH2N(Et)C(O)OκS,O) 5.§ The complex was obtained cleanly as a single diastereomer. In the 1H NMR spectrum, the two S–CH2–N protons are diastereotopic and appear as doublets at δ 2.67 and 3.47 (2JHH 14 Hz) respectively. The CH2 protons of the ethyl group appear as doublets of quartets at δ 2.67 and 3.47 (2JHH 14 Hz, 3JHH 7 Hz).
X-Ray diffraction analysis of a red columnar single crystal of cis-Fe(dmpe)2(SCH2N(Et)C(O)O-κS,O) 5 confirmed its ferracyclic structure, and determined its absolute stereochemistry (Fig. 2).‡ The unit cell contained two crystallographically independent molecules. The six-membered ring is puckered, due in part to the planarity of the carbamate (O–C(O)–N) moiety. The metalloring bond lengths suggest essentially single bonds with a localised carbonyl moiety. Steric interaction between the methyl substituents on the axial phosphorus atoms and the groups which make up the sulfido–carbamate ferracycle most probably determines the configuration at the iron centre leading to the formation of a single diastereomer for this compound.
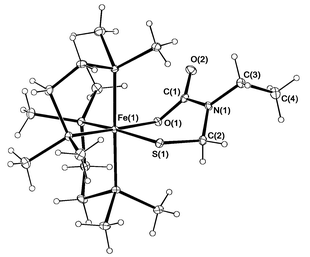 | ||
| Fig. 2 ORTEP5 diagram of one of the molecules of 5 showing 20% displacement ellipsoids. Selected bond distances (Å) and angles (°): Fe(1)–O(1) 2.053(2), C(1)–O(1) 1.286(3), C(1)–O(2) 1.246(3), C(1)–N(1) 1.385(3), N(1)–C(2) 1.446(3), C(2)–S(1) 1.833(3), Fe(1)–S(1) 2.3354(13); Fe(1)–O(1)–C(1) 123.34(18), N(1)–C(2)–S(1) 115.8(2). | ||
cis-Fe(dmpe)2(SCH2N(Et)C(O)OκS,O) 5 is formally the product of hydride migration from the iron(II) centre to the iminium carbon of trans-Fe(dmpe)2(SCHN+(Et)CO2−)H 4 with formation of a six-membered ferracycle. The mechanism of the transformation 4 to 5 probably involves loss of one of the coordinated phosphines, with intramolecular attack by the carboxylate group to eliminate a thioaldehyde. The formation of an intermediate thioaldehyde is speculative and there is no diect spectroscopic evidence for this species. Subsequent addition of the metal hydride to the pendant thioaldehyde would form the cyclic product 5 (Scheme 3).
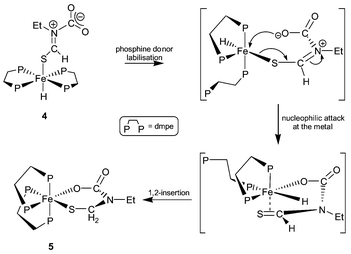 | ||
| Scheme 3 Proposed mechanism for the transformation 4 to 5. | ||
The net result of the overall sequence of reactions is the reduction of the unsaturated isothiocyanate Et–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS to an iron-coordinated, N-substituted thiolate Et–N(X)–CH2–S–Fe by sequential migration of two hydrides from a metal centre to the electrophilic carbon of the heteroallene. CO2 is involved in the reaction initially as an electrophile but is important in the formation of the heterocyclic product. The formation of the carboxylate 4 produces a tethered ligand which is conveniently disposed (via a six-membered transition state) to provide a driving force for the formation of a thioaldehyde and subsequent hydride migration. Further studies of the scope, mechanism and applications of this novel addition/cyclisation reaction of metal hydrides with heteroallenes are ongoing.
CS to an iron-coordinated, N-substituted thiolate Et–N(X)–CH2–S–Fe by sequential migration of two hydrides from a metal centre to the electrophilic carbon of the heteroallene. CO2 is involved in the reaction initially as an electrophile but is important in the formation of the heterocyclic product. The formation of the carboxylate 4 produces a tethered ligand which is conveniently disposed (via a six-membered transition state) to provide a driving force for the formation of a thioaldehyde and subsequent hydride migration. Further studies of the scope, mechanism and applications of this novel addition/cyclisation reaction of metal hydrides with heteroallenes are ongoing.
We thank the Australian Research Council for financial support and the Australian Commonwealth Government for an Australian Postgraduate Award (W. J. S.).
Notes and references
- W. Leitner, Coord. Chem. Rev., 1996, 153, 257 CrossRef CAS.
- L. D. Field, E. T. Lawrenz, W. J. Shaw and P. Turner, Inorg. Chem., 2000, 39, 5632 CrossRef CAS.
- L. D. Field, W. J. Shaw and P. Turner, Organometallics, 2001, 20, 3491 CrossRef CAS.
- D. J. Darensbourg and A. Rokicki, Organometallics, 1982, 1, 1685 CrossRef CAS.
- C. K. Johnson, ORTEP II, Report ORNL-5138, Oak Ridge National Laboratories, Oak Ridge, TN, 1976..
- Bruker, SMART, SAINT and XPREP: Area detector control and data integration and reduction software; Bruker Analytical X-ray Instruments, Inc., Madison, WI, 1995..
- MolecularStructureCorporation teXsan for Windows: Single Crystal Structure Analysis Software, MSC: 3200 Research Forest Drive, The Woodlands, TX, 1997–1998..
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- P. Coppens, L. Leiserowitz and D. Rabinovich, Acta Cryst., 1965, 18, 1035 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33 CrossRef; G. M. Sheldrick, SADABS: Empirical absorption correction program for area detector data, University of Göttingen, Germany, 1996..
- A. Altomare, M. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Cryst., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, SHELXL97: Program for crystal structure refinement, University of Göttingen, Germany, 1997..
Footnotes |
| † Electronic supplementary information (ESI) available: experimental section. See http://www.rsc.org/suppdata/cc/b1/b108492e/ |
‡ Selected crystal data: full sphere data were collected at 150(2) K on a Bruker SMART 1000 to 560 2θ with Mo radiation. Data integration and reduction were undertaken with SAINT and XPREP,6 and subsequent computations were carried out with the teXsan7 and WinGX8 graphical user interfaces. A Gaussian absorption correction was applied to the data.6,9 A subsequent empirical correction was determined with SADABS10 for 4. The data reduction included the application of Lorentz and polarisation corrections. The structures
were solved by direct methods with SIR97,11 and extended and refined with SHELXL-97.124: C16H39FeNO2P4S, M = 489.27, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.973(5), b = 14.680(8), c = 8.748(5) Å, α = 104.586(8), β = 99.791(8), γ = 81.837(8)°, V = 1214.8(11) Å3, Dc = 1.338 g cm−3, Z = 2, R1(F) 0.0391, wR2(F2) 0.1000, GOF(all) 1.059. The equatorial ligand system is disordered with the bidentate ligands adopting two orientations. Complementary populations of the two equatorial ligand orientations were refined and then fixed in both cases. The partially occupied non-hydrogen sites were modelled with isotropic displacement parameters, whereas the rest of
the non-hydrogen sites were treated anisotropically. A riding atom model was used for hydrogen atoms with the exception of H(1) and the hydride H(1Fe), both of which were located and modelled with refined positional and isotropic displacement parameters.5: C16H39FeNO2P4S, M = 489.27, monoclinic, space group P/n (no. 14), a = 17.989(9), b = 12.561(6), c = 20.303(10) Å, β = 94.604(8)°, V = 4573(4) Å3, Dc = 1.421 g cm−3, Z = 8, R1(F) 0.0366, wR2(F2) 0.0489, GOF(all) 0.983. The asymmetric unit contains two crystallographically independent molecules. The non-hydrogen atoms were modelled with anisotropic displacement parameters and a riding atom model was used for the hydrogen atoms. CCDC reference numbers 169564 and 169565. See http://www.rsc.org/suppdata/cc/b1/b108492e/
for crystallographic data in CIF or other electronic format. (no. 2), a = 9.973(5), b = 14.680(8), c = 8.748(5) Å, α = 104.586(8), β = 99.791(8), γ = 81.837(8)°, V = 1214.8(11) Å3, Dc = 1.338 g cm−3, Z = 2, R1(F) 0.0391, wR2(F2) 0.1000, GOF(all) 1.059. The equatorial ligand system is disordered with the bidentate ligands adopting two orientations. Complementary populations of the two equatorial ligand orientations were refined and then fixed in both cases. The partially occupied non-hydrogen sites were modelled with isotropic displacement parameters, whereas the rest of
the non-hydrogen sites were treated anisotropically. A riding atom model was used for hydrogen atoms with the exception of H(1) and the hydride H(1Fe), both of which were located and modelled with refined positional and isotropic displacement parameters.5: C16H39FeNO2P4S, M = 489.27, monoclinic, space group P/n (no. 14), a = 17.989(9), b = 12.561(6), c = 20.303(10) Å, β = 94.604(8)°, V = 4573(4) Å3, Dc = 1.421 g cm−3, Z = 8, R1(F) 0.0366, wR2(F2) 0.0489, GOF(all) 0.983. The asymmetric unit contains two crystallographically independent molecules. The non-hydrogen atoms were modelled with anisotropic displacement parameters and a riding atom model was used for the hydrogen atoms. CCDC reference numbers 169564 and 169565. See http://www.rsc.org/suppdata/cc/b1/b108492e/
for crystallographic data in CIF or other electronic format. |
| § Selected spectral data for 5: 31P{1H} NMR: δ 45.3 (ddd, 2JPP 32, 40, 226 Hz), 57.9 (ddd, 2JPP = 36, 52, 226 Hz), 60.3 (dt, 2JPP 28, 36 Hz), 65.2 (dt, 2JPP 28, 52 Hz). 13C{1H}: δ 45.9 (m, JCP 3, 5 Hz, FeS–CH2–N), 166.1 (p, 3JCP
3 Hz, FeO(CO)N). |
| This journal is © The Royal Society of Chemistry 2002 |