Total synthesis and determination of the stereochemistry of 2-amino-3-cyclopropylbutanoic acid, a novel plant growth regulator isolated from the mushroom Amanita castanopsidis Hongo
Yoshiki
Morimoto
*a,
Mamoru
Takaishi
a,
Takamasa
Kinoshita
a,
Kazuhiko
Sakaguchi
b and
Kozo
Shibata†
a
aDepartment of Chemistry, Graduate School of Science, Osaka City University, Sumiyoshi-ku, Osaka, 558–8585, Japan.. E-mail: morimoto@sci.osaka-cu.ac.jp
bDepartment of Material Science, Graduate School of Science, Osaka City University, Sumiyoshi-ku, Osaka, 558–8585, Japan
First published on 14th December 2001
Abstract
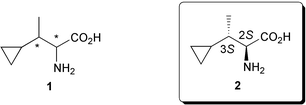
The unknown stereostructure of 2-amino-3-cyclopropylbutanoic acid 1, a novel plant growth regulator isolated from the mushroom Amanita castanopsidis Hongo, was determined to be (2S,3S)-2 through its racemic and enantioselective syntheses employing the chelate–enolate Claisen rearrangement as a key step.
2-Amino-3-cyclopropylbutanoic acid 1 was isolated as a novel α-amino acid from the mushroom Amanita castanopsidis Hongo (Koshiroonitake in Japanese) by Yoshimura et al. in 1999, and the plane structure was elucidated by spectroscopic analyses.1 The relative and absolute configurations at the two chiral centres C2 and C3 have not, however, been determined. It is reported that 1 substantially inhibits root elongation in lettuce seedlings, and the biological activity is like indole-3-acetic acid known as an important plant growth regulator. There have also been many cyclopropane ring-containing α-amino acids closely related to 1, such as coronatine,2 cyclopropylalanine,3 methylenecyclopropylglycine and hypoglycin A4 and so forth, possessing a broad spectrum of biological activities. Thus, it appeared desirable to clarify the stereostructure of 1 for exploring structure–activity relationships between the cyclopropane ring-containing α-amino acids mentioned above and 1. In this paper, we report that the stereochemistry of 1 is 2S,3S through the stereoselective syntheses of two possible syn and anti diastereomers 7a and 7b, respectively, in racemic form and the optically active anti2.
First, we decided to synthesize two possible diastereomers, syn7a and anti7b, to elucidate the unknown relative configuration. In 1994, it has been reported by Kazmaier that syn amino acid 4a can diastereoselectively be obtained in a ratio of syn∶anti = 95∶5 from (E)-ester 3avia the [3,3]-sigmatropic rearrangement of a chelate-bridged zinc enolate (Scheme 1).5 The high diastereoselectivity was explained in terms of a chairlike transition state A (R1 = RZ = H, R2 = Z, RE = Me) like that generally postulated for [3,3]-sigmatropic rearrangements of acyclic systems (Scheme 2).6 Therefore, another anti amino acid 4b could be constructed by applying Kazmaier’s method to (Z)-ester 3b.
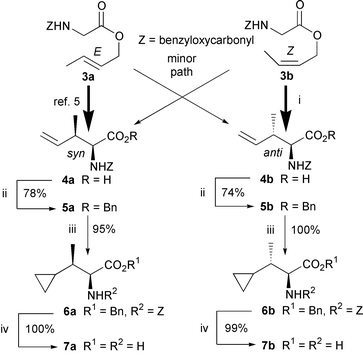 | ||
| Scheme 1 Reagents and conditions: i, LDA, ZnCl2, THF, −78 °C to RT, 30 min, 47%; ii, DCC, DMAP, BnOH, Et2O, RT, 20 h; iii, CH2N2, Pd(OAc)2, Et2O, 0 °C, 1 h; iv, H2, 10% Pd–C, MeOH, RT, 15 min. | ||
![Plausible transition states for the [3,3]-sigmatropic rearrangement of chelate-bridged zinc enolates.](/image/article/2002/CC/b108752e/b108752e-s2.gif) | ||
| Scheme 2 Plausible transition states for the [3,3]-sigmatropic rearrangement of chelate-bridged zinc enolates. | ||
In practice, the [3,3]-sigmatropic rearrangement of (Z)-ester 3b‡ under Kazmaier’s conditions predominantly afforded the anti amino acid 4b7 in a modest yield (anti∶syn = ca. 80∶20). The somewhat lowered diastereoselectivity in the rearrangement of (Z)-3b may be due to the following reason. In the case of (Z)-3b, a steric repulsion between the axial substituent RZ and the rigid chelate-bridged zinc enolate moiety in the transition state A (R1 = RE = H, R2 = Z, RZ = Me) leading to the anti4b is more serious than in that of (E)-3a. Therefore, since the energy difference between A and the boatlike transition state B leading to the syn4a becomes smaller than that in (E)-3a, the lowering of the diastereoselectivity might be observed. Anyway, both the diastereomers 4a and 4b were converted into the desired syn and anti 2-amino-3-cyclopropylbutanoic acids 7a and 7b, respectively, by a sequence of reactions: (i) protection of the carboxyl group as a benzyl ester; (ii) Pd(OAc)2-catalyzed cyclopropanation of the terminal double bond with diazomethane;8 and (iii) hydrogenolysis of the benzylic protective groups. Comparing the 1H and 13C NMR of synthetic 7a and 7b with those of the natural product 1,1 it has been found that 1 is identical to 7b possessing the anti relative stereochemistry.
Next, we embarked on the enantioselective synthesis of optically active anti2 bearing 2S stereochemistry, because Yoshimura et al. have deduced that the absolute configuration at the C2 position of 1 is S.1 Recently, Sakaguchi et al., one of the authors in this paper, have developed silyl-assisted [3,3]-sigmatropic rearrangements of (1-acyloxy-2-alkenyl)trialkylsilanes to provide optically active vinylsilane-containing α-amino acids in a complete chirality-transferring manner.9 In conjunction with the key step for the syntheses of racemates 7a and 7b, we adopted the method to synthesize the optically active 2.
The preparation of the rearrangement precursor (Z)-13 (J = 11.0 Hz between the olefinic protons) was carried out by esterfication of the readily available chiral (S)-alcohol 11,10 [α]23D −90.2 (c 1.01, CHCl3); 98% ee, with commercially available Boc-glycine followed by partial reduction of the resulting alkyne 12 with Lindlar catalyst (Scheme 3). The [3,3]-sigmatropic rearrangement of 13 diastereo- and enantioselectively proceeded under Kazmaier’s conditions to give only the (E)-anti product 14 (J = 18.5 Hz between the olefinic protons) in 97% yield with complete chirality transfer.§ In spite of the same (Z)-geometry as 3b, the high diastereo- and enantioselectivity observed in this rearrangement could be attributed to the bulky tert-butyldimethylsilyl group. In the transition state B (R1 = TBDMS, R2 = Boc, RZ = Me, RE = H) leading to syn8 and C leading to 10 enantiomeric to 9 except for the alkene geometry, sterically encumbered A1,3-strain and 1,3-diaxial-like interaction, respectively, occur between the bulky R1 and RZ. Therefore, because the transition state A without such a repulsive interaction becomes relatively more stable than B and C, 14 might exclusively be formed.
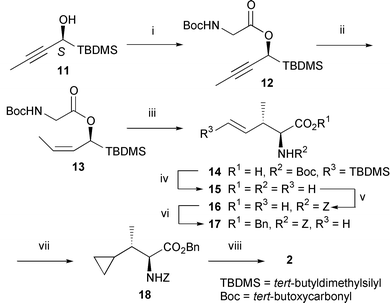 | ||
| Scheme 3 Reagents and conditions: i, Boc-glycine, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI), DMAP, CH2Cl2, RT, 2 h, 94%; ii, H2, 5% Pd–CaCO3, Py, EtOAc, RT, 1 h, 81%; iii, LDA, ZnCl2, THF, −78 °C to RT, 2 h, 97%; iv, 42% aq. HBF4, 1,4-dioxane, 65 °C, 14 h, 100%; v, ZCl, K2CO3, 1,4-dioxane–H2O (1∶1), 0 °C to RT, 12 h, 73%; vi, BnOH, EDCI, DMAP, CH2Cl2, RT, 4 h, 94%; vii, iii in Scheme 1, 100%; viii, iv in Scheme 1, 100%. | ||
Concurrent removal of the TBDMS and Boc groups in the rearrangement product 14 with 42% HBF411 furnished free amino acid 15, N-Z protection of which yielded 16 consistent with (±)-4b. Finally, according to the racemic route, the carboxylic acid 16 was converted into the amino acid (2S,3S)-2 in almost quantitative yield. The optical rotation of synthetic 2, [α]31D −9.08 (c 0.5, H2O), was identical with that reexamined for the natural amino acid 1 generously gifted by Professor Wakabayashi (Osaka City University), [α]26D −9.46 (c 0.035, H2O). Thus, it has been found that the hitherto unknown absolute configuration of 1 is assigned to 2S,3S.
In conclusion, we have accomplished the stereoselective syntheses of two possible syn and anti diastereomers 7a and 7b, respectively, in racemic form and the optically active anti2 using the chelate–enolate Claisen rearrangement as a key step, and determined the relative and absolute configurations of 2-amino-3-cyclopropylbutanoic acid 1, a novel plant growth regulator. The elucidation of the stereochemistry of 1 will be useful for structure–activity relationships and conformational analysis.
We thank Professor K. Wakabayashi (Osaka City University) for generously supplying natural amino acid 1, and are also grateful to Ms H. Yoshimura and Mr H. Suzuki (Osaka City University) for helpful discussions.
Notes and references
- H. Yoshimura, K. Takegami, M. Doe, T. Yamashita, K. Shibata, K. Wakabayashi, K. Soga and S. Kamisaka, Phytochemistry, 1999, 52, 25 CrossRef CAS.
- A. Ichihara and H. Toshima, in Biologically Active Natural Products: Agrochemicals, ed. H. Cutler and S. Cutler, CRC Press, Washington, DC, 1999, p. 93. Search PubMed.
- T. Ohta, S. Nakajima, Z. Sato, T. Aoki, S.-I. Hatanaka and S. Nozoe, Chem. Lett., 1986, 511 CAS.
- D. Li, G. Agnihotri, S. Dakoji, E. Oh, M. Lantz and H.-W. Liu, J. Am. Chem. Soc., 1999, 121, 9034 and references cited therein. Search PubMed.
- U. Kazmaier, Angew. Chem., Int. Ed. Engl., 1994, 33, 998 CrossRef.
- P. Wipf, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 5, p. 827 and references cited therein Search PubMed; U. Kazmaier, Liebigs Ann./Recueil, 1997, 285 Search PubMed.
- P. A. Bartlett and J. F. Barstow, J. Org. Chem., 1982, 47, 3933 CrossRef CAS.
- M. Suda, Synthesis, 1981, 714 CrossRef CAS.
- K. Sakaguchi, H. Suzuki and Y. Ohfune, Chirality, 2001, 13, 357 CrossRef CAS.
- K. Sakaguchi, M. Fujita, H. Suzuki, M. Higashino and Y. Ohfune, Tetrahedron Lett., 2000, 41, 6589 CrossRef CAS.
- R. E. Ireland and M. D. Varney, J. Am. Chem. Soc., 1984, 106, 3668 CrossRef CAS.
Footnotes |
| † Deceased on June 18, 1998. |
| ‡ The (Z)-ester 3b (J = 10.8 Hz between the olefinic protons) was prepared by condensation of commercially available Z-glycine with but-2-yn-1-ol (DCC, DMAP, Et2O, RT, 22 h, 97%) and subsequent Lindlar reduction of the resulting alkyne (H2, 5% Pd–CaCO3, Py, EtOAc, RT, 40 min, 89%). All new compounds were satisfactorily characterized using 1H and 13C NMR, IR, MS and HRMS spectra and also by elemental analyses whenever possible. Selected data for 7a: mp 262–264 °C; δH (400 MHz, D2O) 3.74 (1H, d, J = 4.1 Hz), 1.34 (1H, ddq, J = 9.8, 4.1, 7.0 Hz), 0.98 (3H, d, J = 7.1 Hz), 0.68–0.44 (3H, m), 0.25–0.14 (2H, m); δC (100 MHz, D2O) 174.6, 60.1, 39.8, 14.12, 14.10, 4.3, 4.0; νmax (KBr)/cm−1 3600–2200, 3410, 1672; m/z (FAB-MS) 144 [(M + H)+, 100%] (FAB-HRMS: calc. for C7H14O2N [(M + H)+], 144.1025; found, 144.1032) (Calc. for C7H13O2N: C, 58.72; H, 9.15; N, 9.78. Found: C, 58.54; H, 9.30; N, 9.64%). 2: mp 265–268 °C; δH (400 MHz, D2O) 3.64 (1H, d, J = 4.6 Hz), 1.30 (1H, ddq, J = 9.8, 4.6, 7.1 Hz), 1.06 (3H, d, J = 7.1 Hz), 0.65 (1H, m), 0.47 (2H, m), 0.22 (1H, m), 0.09 (1H, m); δC (100 MHz, D2O) 174.3, 60.2, 39.6, 16.1, 12.9, 4.0, 2.7; νmax (KBr)/cm−1 3600–2200, 3342, 1628; m/z (FAB-MS) 144 [(M + H)+, 100%] (FAB-HRMS: calc. for C7H14O2N [(M + H)+], 144.1025; found, 144.1010) (Calc. for C7H13O2N: C, 58.72; H, 9.15; N, 9.78. Found: C, 58.25; H, 9.13; N, 9.55%). |
| § The absolute configuration of (2S,3S)-14, [α]24D +9.94 (c 1.00, CHCl3), has been confirmed by leading the enantiomer of 14, [α]19D –8.6 (c 1.05, CHCl3), to (2R,3R)-D-isoleucine (ref. 9). The optical purity of 14 was determined to be >99% ee by derivatization of 15 (R1 = R2 = R3 = H) to 15′ [R1 = Me, R2 = (R)-α-methoxy-α-(trifluoromethyl)phenylacetyl, R3 = H] and integration of the signals in the1 H NMR spectrum. |
| This journal is © The Royal Society of Chemistry 2002 |