Determination of liposome/water partition coefficients of organic acids and bases by solid-phase microextraction
Beate I.
Escher
*a,
Michael
Berg
a,
Jürg
Mühlemann
a,
Martin A. A.
Schwarz
a,
Joop L. M.
Hermens
b,
Wouter H. J.
Vaes
c and
René P.
Schwarzenbach
a
aSwiss Federal Institute for Environmental Science and Technology (EAWAG) and Swiss Federal Institute of Technology (ETH), CH-8600 Dübendorf, Switzerland. E-mail: escher@eawag.ch; Fax: +41 1 823 5471; Tel: +41 1 823 5068
bInstitute of Risk Assessment Sciences, Division Toxicology, Utrecht University, 3508 TD Utrecht, The Netherlands
cTNO Nutrition and Food Research, 3700 AJ Zeist, The Netherlands
First published on 12th December 2001
Abstract
The extraction of two methylated anilines and three chlorinated phenols by solid-phase microextraction (SPME) fibers coated with polyacrylate was investigated as a function of pH. Only the neutral species of the acids and bases partitioned into the polymer. Extraction kinetics were accelerated for the hydrophobic phenols at pH values around their acidity constant. This is presumably due to a reconstitution of the neutral species in the unstirred aqueous layer adjacent to the polymer surface by the charged species through the fast acid–base equilibrium. Although the charged species is not taken up into the polymer, liposome/water distribution ratios could be measured up to a pH value, where 99% of the compounds were present as charged species. The partition coefficients of the neutral and charged species were extrapolated from the pH profiles of the liposome/water distribution ratios. The resulting values were slightly lower than those measured with equilibrium dialysis. The discrepancies are discussed with respect to differences in the experimental conditions and the possibility of matrix effects during SPME measurements.
Introduction
Solid-phase microextraction (SPME) is a solvent-free extraction technique that has many technical advantages, in particular convenience, since it can be directly coupled to gas chromatography (GC), where the analyte is thermally desorbed in the injector port,1 or to high-performance liquid chromatography (HPLC) via a special interface.2 Besides its application as an analytical method for the quantitative determination of organic pollutants and drugs in aqueous samples or in the headspace of aqueous samples,3–5 SPME is increasingly being used to perform biomimetic extractions and to assess the bioavailability of organic pollutants in the presence of organic matrices.6–11 Both strategies rely upon the premise that only the free aqueous concentration of a pollutant is available for biouptake and for partitioning into the polymer coating of the SPME fiber. Since partitioning to hydrophobic fiber coatings is, at least for apolar compounds, proportional to partitioning into organic solvents that mimic biological tissues (e.g., octanol),12 and hence to bioconcentration,13 SPME has been used as tool for biomimetic extractions14 and as a surrogate for total body residues of complex environmental mixtures.15As a prerequisite for the use of SPME as a method to determine free aqueous concentrations, partitioning to the SPME fiber should not disturb the equilibrium between the free aqueous concentration and the concentration in the organic or biological matrix. This condition can be achieved by the so-called negligible depletion SPME (nd-SPME) technique introduced by Vaes et al.6,16 For nd-SPME, less than 5% of the analyte should be extracted and the organic matrix should not interact with the SPME fiber. This method has been successfully applied to the determination of the partitioning of pollutants from water to proteins,6,17 liposomes,18 blood and other tissues from rats,19 and dissolved organic material.7–9,20 In addition, SPME can be used to measure the aqueous solubilities and octanol/water partition coefficients.21,22
Interactions between the organic matrix and the SPME fiber have been shown to be negligible for the protein bovine serum albumin6 and for humic acid (Aldrich).7 Proteins from human blood serum were found to interact with the SPME fiber, causing problems with reproducibility and fiber lifetime.23 Such interactions may lead to complications and artifacts, primarily, for very hydrophobic compounds due to the influence of the matrix on the uptake kinetics.24
SPME has also been used in the analysis of ionogenic compounds, like weak organic acids and bases. Usually, the emphasis was put on optimizing the extraction efficiency by adjusting the pH so that the compound was in its neutral speciation form25,26 but some studies worked at ambient pH values or varied the pH. In most studies, it was assumed that only the neutral species is partitioning into the polymer coating of the SPME fiber.9,25,27 However, Holten Lützhøft et al.28 showed that partitioning of the positively and negatively charged species of 4-quinolones was about one order of magnitude smaller than the corresponding values for the neutral species, but were not negligible.
The aim of the present study was to evaluate the applicability of nd-SPME for the determination of pH profiles of liposome/water partitioning of weak organic acids (chlorophenols) and weak organic bases (methylanilines). For ionogenic compounds, partitioning into biological material depends on the speciation and hence for acids and bases it depends on the pH. For the investigated set of compounds, it was shown that the neutral species exhibits about a ten-fold higher partitioning coefficient than the charged species (protonated aniline or deprotonated phenol).29 In contrast, partitioning of the charged species into organic bulk solvents is negligible (e.g., in alkane/water systems) or only possible if ion pairs are formed in the organic phase (e.g., in the octanol/water system).30 Therefore bulk solvents cannot be used as surrogates for biomembranes for use with ionogenic compounds but screening methods have to be developed for the determination of liposome/water partition coefficients. Established methods include equilibrium dialysis, pH-metric titrations and the use of lipid bilayers immobilized on solid support material.31 The method of nd-SPME is a possible alternative for these methods but has not yet been applied for determining the pH dependence of liposome/water partitioning of organic acids and bases.
Experimental
Chemicals
The compounds (names and abbreviations are given in Table 1) were purchased from the following companies: 26DCP, 246TriCP, 2346TeCP, Riedel-de Haën (Seelze, Germany); PCP, 246TMA, CHI, TOL, Fluka AG (Buchs, Switzerland); 34DMA, DIB, Aldrich (Buchs, Switzerland). All chemicals were of highest purity available (=97%) and were used as received. Egg yolk lecithin was purchased from Lipoid (Ludwigshafen, Germany). The particular preparation of lecithin used contained =98% phosphatidylcholine and its overall fatty acid ester composition was 30–33% palmitate, 11–14% stearate, 29–32% oleate, 14–16% linoleate, with minor fractions of other fatty acids esters. All pH buffers were purchased from Fluka: phosphate, acetate, MES (2-morpholinoethanesulfonic acid, pKa = 6.15), MOPS (3-(N-morpholino)propanesulfonic acid, pKa = 7.2); CHES (2-(cyclohexylamino)ethanesulfonic acid, pKa = 9.55).| Compound | Abbreviation | pKa | LogKow (neutral) | LogDfw,n(t) |
|---|---|---|---|---|
| a Data taken from ref. 29. b PA fiber, 85 μm, Ctot = 100 μM, 40 min extraction at 30 °C. c Experimental value determined at pH 7.4. d Data taken from ref. 32. e Data taken from ref. 33. f Data taken from ref. 34. g Average of experimental values at all pH values. h PA fiber, 85 μm, Ctot = 100 μM, 10 min extraction at 30 °C. i Experimental value determined at pH 2. | ||||
| 3,4-Dimethylaniline | 34DMA | 5.23a | 1.87a | 1.49bc |
| 2,4,6-Trimethylaniline | 246TMA | 4.38a | 2.35a | 1.85bc |
| Chinoline | CHI | 4.90d | 2.03e | 1.63bc |
| Toluene | TOL | — | 2.69f | 1.96bg |
| 2,6-Dichlorophenol | 26DCP | 6.97d | 2.64a | 2.6hi |
| 2,4,6-Trichlorophenol | 246TriCP | 6.15d | 3.72a | 2.89hi |
| 2,3,4,6-Tetrachlorophenol | 2346TeCP | 5.40a | 4.42a | 2.91hi |
| Pentachlorophenol | PCP | 4.75a | 5.24a | 2.93hi |
| 1,4-Diiodobenzene | DIB | — | 4.11e | 2.51gh |
Analytical method
SPME devices with 10 mm fibers coated with 85 μm polyacrylate (PA) and 30 and 100 μm polydimethylsiloxane (PDMS) were purchased from Supelco (Bellefonte, PA, USA). For some experiments with liposomes, the fibers were cut to 1–2 mm to meet the requirement of negligible depletion extraction. The PA fibers were conditioned for 2 h at 300 °C under a stream of hydrogen, the PDMS fibers for 1 h at 250 °C. The PA fiber has been widely recommended for use in the analysis of phenols and of other aromatic acids and bases,9,25,26 while the less polar PDMS fiber is generally used for more hydrophobic analytes. As preliminary tests showed (data not shown), the PDMS fiber has a lower extraction efficiency as compared with the PA fiber, a slight carry-over, and fiber-bleaching peaks in the gas chromatogram. Therefore, in the following, only the PA fiber was used.For the extraction and desorption process, a CTC CombiPAL autosampler equipped for SPME was used (CTC Analytics, Zwingen, Switzerland). All extractions were performed at 30 °C and the vials were vibrated to accelerate the extraction kinetics. All analyses were performed with a Fisons 8165 GC (Fisons Instruments, Manchester, UK) equipped with a split/splitless injector, a 30 m × 0.25 mm fused silica DB-5 MS column (J&W Scientific Folson, CA, USA) or a 30 m × 0.25 mm BPX5 (SGE, Ringwood, Australia), both with a 0.25 μm film thickness and a 1 m deactivated pre-column (BGB Analytik, Anwil, Switzerland). An electron capture detector, ECD 80, was used for the analysis of the chlorinated phenols, and a flame ionization detector, FID 80, module (both Fisons Instruments, Manchester, UK) was used for other compounds.
The injector was kept at 275 °C for the chlorinated phenols and at 270 °C for the other analytes. The split valves were closed for the analysis, but opened fully during fiber conditioning. A custom-made, narrow-bore insert liner with a diameter of 0.75 mm was used for SPME (Supelco, Bellefonte, PA, USA). During the 5 min injection period the oven was kept at 40 °C, after which the temperature was raised at 15 °C min−1. to 275 °C. The oven was then kept at 275 °C for an additional 5 min. Hydrogen of purity 50 was used as carrier gas. The carrier gas flow was kept constant at 1.3 mL min−1. The ECD was operated with nitrogen of purity 57 as make-up gas at a rate of 40 mL min−1. The FID worked with hydrogen, compressed air and N2 at 20 mL min−1 as make-up gas.
SPME extraction studies of ionogenic compounds
The samples were prepared by diluting a methanolic stock solution (0.01 M) of a given compound in buffer solution at defined pH values to a concentration of 100 μM. To determine the detector response a defined amount of analyte dissolved in hexane or ethyl acetate was injected with cold on-column injection. Using this method the whole amount of substance injected was supposed to reach the detector and give a response. The thus determined ratio of area counts per mass of analyte was used to calculate the total amount extracted by SPME, nf. The extraction efficiency or fraction of analyte in the fiber, ff, is defined as: | (1) |
 | (2) |
Uptake kinetics were measured at various pH values with extraction times ranging from 5 to 250 min at 30 ± 1 °C. For the liposome/water partitioning experiments, extraction times resulting in <2% extraction efficiency (and in a few exceptional cases <5%) were chosen, and, if the required extraction time was below 10 min, the fiber was cut from 1 cm original length to 1 or 2 mm to meet the requirement of negligible depletion.6 The buffer composition and the variation of pH did not affect the partitioning to the fiber as was shown by one experiment at pH 3.4 with all buffer components used separately and by using neutral reference compounds, DIB or TOL, in all pH dependent experiments.
Liposome/water distribution experiments
Experiments were conducted in two sets of vials. The reference vial contained a given amount of analyte at a given pH, the measurement vial additionally contained a known amount of liposomes. Large unilamellar liposomes made up of phosphatidyl choline were prepared by membrane extrusion as described in ref. 29, where further information on the characterization of the liposomes can be found. The overall liposome concentration was adjusted to an expected 50% depletion of the aqueous phase by uptake into the liposomes. For every pH unit, 4–10 different analyte concentrations (each measured in triplicate) were used to derive sorption isotherms. After equilibration by shaking for 30 min, the free aqueous concentrations in both sets of test vials, Cw,ref for the reference vial and Cw for the measurement vial, were determined with SPME as described above. The liposome/water distribution ratios were deduced from the slopes of the sorption isotherms as given by eqn. (3): | (3) |
Equilibrium dialysis experiment
One equilibrium dialysis experiment was performed with 246TriCP at pH 4. The nominal concentration of 246TriCP was kept constant at 10 μM and the liposome concentration was varied from 3 × 10−5 to 9.2 × 10−4 kg L−1. The experiment was performed according to the procedure described in ref. 29. The contents of both dialysis half-cells (with and without liposomes) were transferred into SPME-autosampler vials and SPME analysis was performed as described above. In parallel, the same experiment was performed by direct SPME analysis as described above but also with constant nominal 246TriCP concentration and variable liposome concentrations. The design of the experiment is visualized in Fig. 1.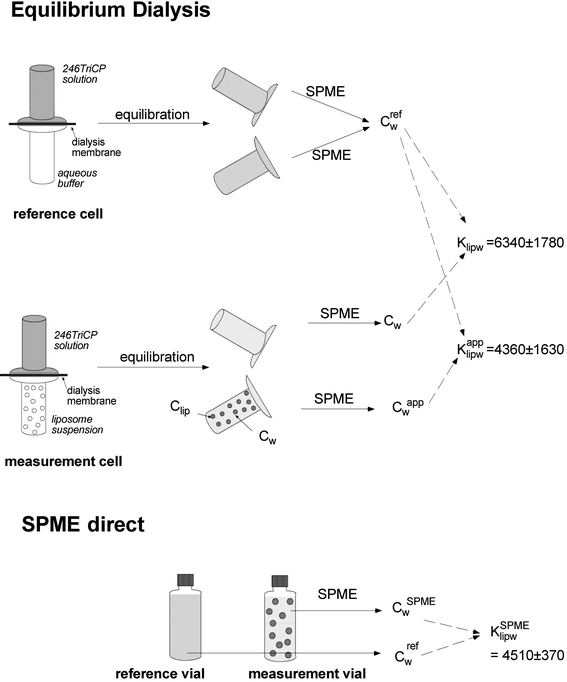 | ||
| Fig. 1 Experimental set-up and overview of results for the combined equilibrium dialysis and SPME experiment of 246TriCP at pH 4. | ||
Results and discussion
Extraction kinetics
Although the nd-SPME method requires low extraction efficiencies and is usually not performed as equilibrium extraction,6 extraction kinetics of the acids and bases were investigated in order to characterize the influence of pH and diffusion limitations, and to find the optimum measurement conditions for the liposome/water partition experiments.Extraction kinetics were measured as a function of pH at 30 °C. As depicted in Fig. 2A for 34DMA and in Fig. 2C for 246TriCP, the extraction equilibrium was not fully reached, even after 250 min of extraction. Extraction efficiency increased for the base 34DMA with increasing pH and decreased for the acid 246TriCP with increasing pH. If it is assumed that only the neutral species is partitioning into the fiber, the extraction kinetics can be replotted, as is shown in Fig. 2B and D. In both cases, all three uptake curves of a given compound are overlaying each other. This is an indication that the initial assumption is reasonable and that the fiber/water partitioning of the charged species can be neglected. Holten Lützhøft et al.28 performed analogous experiments with 4-quinolones on a carbowax/templated polymer resin fiber. Both the anionic and the cationic species partitioned into this fiber type but the distribution ratio of the charged species is about one order of magnitude smaller than of the neutral species. Such a small difference would not show up in the type of data analysis we performed here.
 | ||
| Fig. 2 A and B: Extraction kinetics of 34DMA determined at 30 °C with an 85 μm PA fiber, length 10 mm; ◆ pH 4.7, ▲ pH 5.2, ● pH 7.2. A: Uptake curve varies with pH. B: Uptake normalized to concentration of neutral species. C and D: Extraction kinetics of 246TriCP determined at 30 °C with an 85 μm PA fiber cut to 1 mm; ◆ pH 4.7, ▲ pH 6.2, ● pH 7.2. C: Uptake curve varies with pH. D: Uptake normalized to concentration of neutral species. The solid lines are fits of the experimental data to eqn. (5). | ||
For compounds of low hydrophobicity, the rate of uptake is limited by the diffusion of the compound through the polymer phase.16 In this case, the uptake kinetics should not be influenced by the organic material or other matrices present. Indeed, the presence of humic acids did not significantly influence the uptake kinetics of 2,5-dichlorophenol9 and of 4-quinolone oxolinic acid.28 If the diffusive resistance of the fiber is limiting the uptake process, the uptake rate constant, k1, is directly proportional to the fiber/water partition coefficient, Kfw.
In contrast, for more hydrophobic compounds, diffusion through the unstirred aqueous layer becomes rate limiting. In this case, the matrix can influence the uptake kinetics as was shown for hydrophobic PCBs in a soil–chyme matrix by Oomen et al.24 In this case, k1 is approximately constant and the elimination rate constant, k2, decreases with increasing hydrophobicity. Measurements at low extraction times will therefore give extraction yields independent of the hydrophobicity of the compounds.
For the two compounds of low hydrophobicity, 34DMA and 246TMA, uptake is fast enough that the steady state Dfw can be derived from the uptake kinetics with the following first-order, one-compartment model.16
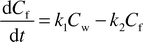 | (4) |
 | (5) |
The uptake rate constant k1 was 2.67 ± 0.29 min−1 for 34DMA and 12.1 ± 0.9 min−1 for 246TMA, while the elimination rate constants were indistinguishable, with a value of 0.016 ± 0.003 min−1 and 0.015 ± 0.002 min−1 for 34DMA and 246TMA, respectively. These values are in agreement with the data of neutral compounds presented by Verbruggen et al.,15 confirming that the kinetics are rate limited by the diffusion in the fiber.
For the chlorinated phenols, uptake is even slower, with an almost linear uptake curve for 2346TeCP and PCP (data not shown). As it is the case for the anilines, for 246TriCP, the pH dependent uptake curves collapse into a single curve if the uptake of the anionic phenoxide species is assumed to be negligible (Fig. 2D). 246TriCP might be a borderline case between limitation by the polymer and by the aqueous phase but 2346TeCP and PCP are clearly diffusion limited by the aqueous phase. This is indicated by the fact that the extraction efficiencies after 10 min of extraction were equal for all the more highly chlorinated phenols at pH 2, where only the neutral species is present (Table 1).
Based upon the findings described above, an extraction time of 40 min was chosen for the anilines and an extraction time of 10 min for the phenols. Under these conditions, the pH profiles of all the compounds plus two neutral reference compounds were determined. The invariance of Dfw(pH,t) of the neutral compounds TOL (Fig. 3A) and DIB (Fig. 3B) to changes in pH indicates that the pH dependence is solely due to the speciation of the organic base and not due to the effects of the buffer or changes in the fiber.
 | ||
| Fig. 3 pH dependence of the fiber/water distribution ratio for an 85 μm PA fiber, extraction time 40 min A, Organic bases: ▲ 34DMA, ■ chinoline, ◆ 246TMA, ○ non-ionogenic reference toluene. Drawn lines: models described by eqn. (6). B, Organic acids, PA fiber, extraction time 10 min (● 26DCP, ▲ 246TriCP, ■ 2346TeCP, ◆ PCP, ○ non-ionogenic reference DIB). Broken lines: models described by eqn. (6). The error bars correspond to the 95% confidence interval limits. | ||
If it is assumed that charged species do not partition to the PA polymer, then the pH dependence of Dfw(pH,t) for organic acids and bases can be described by
| Dfw(pH,t) = αnDfw,n(t) | (6) |
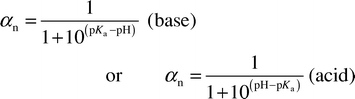 | (7) |
While the bases can be perfectly fitted with eqn. (6), the phenol data cannot be described satisfactorily with the simple model. It appears as if there is a pKa shift to a value around 7 (Fig. 3B). This shift might be an artifact that occurs if the diffusion in the aqueous phase is rate limiting. In this case, the neutral “substrate” for uptake into the fiber could be readily delivered from the conjugate base because the acid–base equilibrium is much faster than the diffusion through the unstirred layer. This hypothesis resembles that put forward by Oomen et al.24 for the uptake of PCBs in the presence of a soil–chyme matrix, where fast desorption of the chemical from the matrix may cause an additional flux to the fiber. In case of the acids such an effect would be particularly pronounced at pH values just above the pKa. This corresponds exactly to the observation in Fig. 3B. A further support of this hypothesis is the air/water exchange of organic acids, where the conductance through the water film is increased at pH values where the charged conjugate base is dominant but it is only the concentration difference of the neutral species between the air and water phase that drives the air/water exchange.35
The most important conclusion from these experiments is that SPME measurements can be conducted over a wider pH range than initially expected, thereby allowing liposome/water partitioning experiments over a wider pH range.
Liposome/water partitioning
Although it was not possible to measure the Klipw values of the charged species directly with the SPME method, it was possible to go up to a fraction of the charged species of 95% for 34DMA, 99% for 246TMA, 92% for 246TriCP, 98% for 2346TeCP and 99.6% for PCP. The apparent Dlipw(pH) were plotted against the fraction of the charged species, as is depicted in Fig. 4, and the partition coefficients of the neutral and charged species, Klipw,HA and Klipw,A for the acids and Klipw,B and Klipw,HB for the bases were extrapolated with eqns. (8) and (9).| Dlipw(pH) = αnKlipw,HA + (1 − αn)Klipw,A | (8) |
| Dlipw(pH) = (1 − αn)Klipw,HB + αnKlipw,B | (9) |
 | ||
| Fig. 4 Plot of the fraction of the charged species versus Dlipw. A, Organic bases: ▲ 246TMA, ■ 34DMA. B, Organic acids: ◆ PCP, ▲ 2346TeCP, ■ 246TriCP. | ||
| SPME | Equilibrium dialysis | |||||
|---|---|---|---|---|---|---|
| Compound | LogKlipw,HA or logKlipw,B | LogKlipw,A or logKlipw,HB | LogKlipw,HA or logKlipw,B | LogKlipw,A or logKlipw,HB | ||
| a Data taken from ref. 29. | ||||||
| 34DMA | 1.98 | 1.67 | 2.11 | 1.99 | ||
| 246TMA | 2.28 | 1.74 | 2.38 | 2.12 | ||
| 246TriCP | 3.70 | 2.32 | 3.99 | 2.50 | ||
| 2346TeCP | 4.26 | 2.60 | 4.46 | 3.46 | ||
| PCP | 4.68 | 3.85 | 5.10 | 4.35 | ||
 | ||
| Fig. 5 Comparison of Klipw determined with the SPME method and the equilibrium dialysis method (Table 2): ■ Klipw,BH, ▲ Klipw,B , ● Klipw,A, ◆ Klipw,HA. Error bars correspond to the 95% confidence interval (for the neutral compounds the error bars are not visible because they are smaller than the size of the symbol). | ||
Equilibrium dialysis is often used as a reference method for the determination of liposome/water partition coefficients,36 although this method is not free of experimental artifacts.37 The most critical artifact in the use of equilibrium dialysis for measuring liposome/water partitioning is the concentration dependent sorption of the analyte to the glass walls and to the dialysis membrane. However, a comparative study with the same test set of compounds has shown good agreement between the equilibrium dialysis method and other experimental methods.31
Nevertheless, there are some differences in the experimental set up of the two methods. The temperature was 30 °C for the SMPE method but 25 °C for the equilibrium dialysis method. Van Wezel et al.38 determined the temperature dependence of the Klipw values of chlorobenzenes and found a slight decrease of Klipw with increasing temperature above the phase-transition temperature from the gel to liquid crystalline states. For the compounds investigated in the present study, an analogous effect might partially account for the observed difference between the two experimental conditions.
There are also small differences in the composition of the acyl chains of the phospholipids but in all cases phosphatidylcholine was used. Earlier work showed that the acyl chain should not have any significant influence on Klipw.29,31
Although great care was taken to exclude saturation of the sorption isotherms, both by choosing appropriate low concentrations of the analytes and by inspection of the linearity of the measured sorption isotherm, for the charged compounds a small artifact due to saturation might account for the difference between the neutral and charged compounds. In conclusion, the described differences in the experimental set-up of the two methods under comparison might fully account for the observed differences in the results.
An additional experiment with combined equilibrium dialysis and SPME was performed with 246TriCP at pH 4 in order to test for artifacts in the SPME measurements in the presence of a matrix. A similar experiment has been conducted for protein/water partitioning and it was found that bovine serum albumin did not sorb to the SPME fiber to such an extent that it increased the concentration in the fiber.6
In the equilibrium dialysis cell, the liposomes are physically retained in one half-cell but the equilibrium between the liposomes is maintained throughout both compartments. Therefore the aqueous concentrations in both half-cells are equal. The aqueous concentrations were determined by SPME from the dialysis half-cell both with and without liposomes and are referred to as Cwapp and Cw, respectively. The experimental set-up and the results are presented in Fig. 1. Cwapp was 10–15% higher than Cw, resulting in a Klipwapp 32% smaller than the Klipw calculated from Cw out of the half-cells without liposomes. The Klipwapp from the equilibrium dialysis experiment was identical to the KlipwSPME determined by the direct SPME method (Fig. 1).
There are two main possibilities to account for this artifact of 10–15% more compound extracted by SPME in the presence of liposomes. First, if diffusion in the aqueous unstirred layer is rate limiting, an extra flux of analyte to the fiber can be induced due to desorption of the analyte from the liposomes. Such an effect has been described for hydrophobic PCBs in the presence of a protein-rich solution but not in the presence of artificial soil.24 As was discussed above, 246TriCP might be a borderline case with regard to diffusion limitation of the extraction process. The higher chlorinated phenols appear to be rate limited by diffusion in the aqueous layer, while the extraction of the less hydrophobic anilines is limited by diffusion in the polymer. For a kinetic effect to be relevant, one should see a larger effect for more hydrophobic molecules than for more hydrophilic ones. Although the data in Fig. 5 show some variation, a slight increase in the difference between the Klipw values of the non-ionized species derived from both techniques can be observed.
The second hypothesis is that liposomes sorb to the SPME fiber and the analyte taken up into the sorbed liposome is quantified by GC after thermal desorption just like analytes sorbed to the fiber polymer. Such observations have not yet been reported for liposomes but human blood plasma was shown to adhere to the PDMS-coated fibers, causing a rapid deterioration of the fiber due to a film of carbonized proteins.23 In the experiments presented here, no irregular deterioration or decrease of the lifetime of the fiber was observed in the presence of liposomes.
From the difference between Cwapp and Cw in the presence of varying liposome concentration, a rough estimate of the amount of liposomes that would be sorbed to the fiber can be calculated under the assumption that the Klipw is identical in free and sorbed liposomes. If about 1 μg of lipids is sorbed to the fiber, which corresponds to 0.05% to 0.8% of the total amount of liposomes in one experiment, the observed 32% difference in apparent partition constant can be explained. Which of the above explanations is valid is difficult to conclude from this study and additional work would be needed to get a complete picture.
Conclusion
SPME has proven to be a good tool to perform extractive analysis of ionogenic compounds even if they are 99% dissociated and the charged species is not taken up into the fiber because the experimental conditions (extraction time and fiber length, i.e., polymer volume) can be adjusted to optimize the conditions between negligible depletion and detection limit. Slightly lower membrane/water partition coefficients are observed using the nd-SPME technique in comparison with the values derived from the dialysis experiments. Potential causes for these differences can be either an influence of the matrix on the kinetics or binding of the lipids to the SPME fiber. The advantages of SPME are its simplicity and it is much less tedious than equilibrium dialysis. However, before taking profit of these advantages, potential matrix effects should be studied carefully. Alternative options are to apply headspace instead of direct immersion SPME and to measure at equilibrium,39 although such an option is applicable only to more volatile chemicals and chemicals that are not too hydrophobic because the equilibration time will otherwise become extremely long. Also the application of other materials that show faster kinetics, such as the application of thin films40 may be a promising alternative. SPME has great potential in estimating the freely dissolved, bioavailable concentration of a compound in a complex matrix (e.g., in an in vitro bioassay), if an appropriate extraction fiber is used and if quality control experiments are conducted to quantify potential matrix effects.Acknowledgement
We thank Bianca Wisner for performing the equilibrium dialysis experiment.References
- C. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145 CrossRef CAS.
- J. Chen and J. Pawliszyn, Anal. Chem., 1995, 67, 2530 CrossRef CAS.
- Applications of Solid Phase Microextraction, ed. J. Pawliszyn, Royal Society of Chemistry, Cambridge, 1999 Search PubMed.
- S. Ulrich, J. Chromatogr., A, 2000, 902, 167 CrossRef CAS.
- H. D. Yuan and J. Pawliszyn, Anal. Chem., 2001, 73, 4410 CrossRef CAS.
- W. H. J. Vaes, E. Urrestarazu-Ramos, H. J. M. Verhaar, W. Seinen and J. L. M. Hermens, Anal. Chem., 1996, 68, 4463 CrossRef CAS.
- E. Urrestarazu-Ramos, S. N. Meijer, W. H. J. Vaes, H. J. M. Verhaar and J. L. M. Hermens, Environ. Sci. Technol., 1998, 32, 3430 CrossRef.
- J. Poerschmann, Z. Y. Zhang, F. D. Kopinke and J. Pawliszyn, Anal. Chem., 1997, 69, 597 CrossRef CAS.
- G. Ohlenbusch, M. U. Kumke and F. H. Frimmel, Sci. Total Environ., 2000, 253, 63 CrossRef CAS.
- D. Sijm, R. Kraaij and A. Belfroid, Environ. Pollut., 2000, 108, 113 CrossRef CAS.
- J. L. M. Hermens, A. P. Freidig, E. Urrestarazu Ramos, W. H. J. Vaes, W. M. G. M. van Loon, E. M. J. Verbruggen and H. J. M. Verhaar, ACS Symp. Ser., 2001, 773, 64 Search PubMed.
- J. R. Dean, W. R. Tomlinson, V. Makovskaya, R. Cumming, M. Hetheridge and M. Comber, Anal. Chem., 1996, 68, 130 CrossRef CAS.
- M. Nendza, in Bioaccumulation in Aquatic Systems, ed. R. Nagel and R. Loskill, VCH, Weinheim, 1991, pp. 43–66 Search PubMed.
- T. F. Parkerton, M. A. Stone and D. J. Letinski, Toxicol. Lett., 2000, 112, 273 CrossRef.
- E. M. J. Verbruggen, W. H. J. Vaes, T. F. Parkerton and L. M. Hermens, Environ. Sci. Technol., 2000, 34, 324 CrossRef CAS.
- W. H. J. Vaes, C. Hamwijk, E. Urrestarazu-Ramos, H. J. M. Verhaar and J. L. M. Hermens, Anal. Chem., 1996, 68, 4458 CrossRef CAS.
- M. Abdel Rehim, G. Carlsson, M. Bielenstein, T. Arvidsson and L. G. Blomberg, J. Chromatogr. Sci., 2000, 38, 458 Search PubMed.
- W. H. J. Vaes, E. Urrestarazu-Ramos, C. Hamwick, I. van Holstein., B. J. Blaauboer, W. Seinen, H. J. M. Verhaar and J. L. M. Hermens, Chem. Res. Toxicol., 1997, 10, 1067 CrossRef CAS.
- E. Artola Garicano, W. H. J. Vaes and J. L. M. Hermens, Toxicol. Appl. Pharmacol., 2000, 166, 138 CrossRef CAS.
- J. Poerschmann, T. Gorecki and F. D. Kopinke, Environ. Sci. Technol., 2000, 34, 3824 CrossRef CAS.
- A. Paschke, P. Popp and G. Schüürmann, Fresenius' J. Anal. Chem., 1998, 360, 52 CrossRef CAS.
- A. Paschke, P. Popp and G. Schüürmann, Fresenius' J. Anal. Chem., 1999, 363, 426 CrossRef CAS.
- K. Poon, P. Lam and M. Lam, Chemosphere, 1999, 39, 905 CrossRef CAS.
- A. G. Oomen, P. Mayer and J. Tolls, Anal. Chem., 2000, 72, 2802 CrossRef CAS.
- H. van Doorn, C. B. Grabanski, D. J. Miller and S. B. Hawthorne, J. Chromatogr., A, 1998, 829, 223 CrossRef CAS.
- M. Guidotto, G. Ravaioli and M. Vitali, J. High Resolut. Chromatogr., 1999, 22, 427 CrossRef.
- M. Abdel Rehim, M. Bielenstein and T. Arvidsson, J. Microcolumn Sep., 2000, 12, 308 CrossRef CAS.
- H.-C. Holten Lützhøft, W. Vaes, A. Freidig, B. Halling-Sørensen and J. Hermens, Environ. Sci. Technol., 2000, 34, 4989 CrossRef.
- B. I. Escher, R. P. Schwarzenbach and J. C. Westall, Environ. Sci. Technol., 2000, 34, 3954 CrossRef CAS.
- C. T. Jafvert, J. C. Westall, E. Grieder and R. P. Schwarzenbach, Environ. Sci. Technol., 1990, 24, 1795 CAS.
- B. I. Escher, R. P. Schwarzenbach and J. C. Westall, Environ. Sci. Technol., 2000, 34, 3962 CrossRef CAS.
- D. Perrin, Dissociation Constants of Organic Bases in Aqueous Solution, Butterworths, London, 1965 Search PubMed.
- C. Hansch, A. Leo and D. Hoekman, Exploring QSAR. Hydrophobic, Electronic and Steric Constants, ACS, Washington, DC, 1995 Search PubMed.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, Wiley, New York, 1993 Search PubMed.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, Wiley, New York, 2nd edn., in preparation. Search PubMed.
- S. Krämer, in Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies, ed. B. Testa, V. Waterbeemd, G. Folkers and R. Guy, Verlag Helvetica Chimica Acta, Zürich, Switzerland, 2001, pp. 401–428 Search PubMed.
- J. Oravcova, B. Böhs and W. Lindner, J. Chromatogr., B, 1996, 677, 1 CrossRef.
- A. P. van Wezel, G. Cornelissen, J. K. van Miltenburg and A. Opperhuizen, Environ. Toxicol. Chem., 1996, 15, 203 CAS.
- P. Mayer, W. H. J. Vaes, F. Wijnker, K. C. H. M. Legierse, R. Kraaij, J. Tolls and J. L. M. Hermens, Environ. Sci. Technol., 2000, 34, 5177 CrossRef CAS.
- J. Wilcockson and F. A. P. C. Gobas, Environ. Sci. Technol., 2001, 35, 1425 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |