DOI:
10.1039/B107781C
(Paper)
Analyst, 2002,
127, 49-53
Received
28th August 2001
, Accepted 8th November 2001
First published on 10th December 2001
Abstract
Solid-phase microextraction (SPME) is used as a sample preparation strategy for gas chromatographic (GC) analysis of the seleno amino acids, selenomethionine (SeMet), selenoethionine (SeEt) and selenocystine (SeCys). Acylation of the amino group and esterification of the carboxylic group in these compounds was performed with isobutylchloroformate to increase volatility. The amino acid derivatives were then extracted by silica fibers with polydimethylsiloxane (PDMS) coatings prepared by the sol–gel process. Investigations of extraction time, acid and salt addition, and polymer length (for the sol–gel process) were conducted with the goal of procedural optimization. Initial characterizations were conducted using gas chromatography with flame ionization detection (GC-FID). Inductively coupled plasma mass spectrometric detection was employed for final selenium detection. Sub-ppb detection limits were obtained for all analytes although relative standard deviations were
higher than those typically obtained in solid-phase microextraction.
Introduction
Selenium in plants and animals can replace the sulfur atom of the amino acids. Furthermore, dietary studies have shown that selenium in the form of amino acids is absorbed more readily than selenium in inorganic forms.1 Numerous analytical methods for the analysis of seleno amino acids have utilized high-performance liquid chromatography (HPLC) coupled with element-specific detection.2 However, when utilizing element-specific detection, specifically inductively coupled plasma mass spectrometry, gas chromatography as the separation technique presents several potential advantages. Primarily, this separation technique offers lower limits of detection because 100% of the column effluent is introduced into the plasma as compared with approximately a few percent when standard nebulizers are used for the sample introduction of the HPLC effluent. Additionally, the ICP does not desolvate or vaporize the analytes, thus providing more
complete atomization and ionization. Furthermore, the decreased solvent load on the plasma has been noted to increase its stability and reduce noise levels.3
Analysis of seleno amino acids by gas chromatography requires derivatization of the carboxylic acid and amino groups in order to increase volatility.4 One derivatization approach features simultaneous esterification and acylation of the amino acids by the addition of an alkyl chloroformate.5 An advantage of this derivatization reaction is that it can take place in the aqueous phase, which effectively facilitates the use of solid-phase microextraction (SPME).
Research is underway to investigate the coupling of derivatization reactions and solid-phase microextraction.6 SPME is a relatively new sample preparation technique, which allows preconcentration and extraction to be performed in one step.7 A silica fiber coated with a polymeric substance of choice is exposed to an aqueous sample or headspace for a specific length of time. During sampling, analyte concentrations reach equilibrium between the aqueous phase and the fiber coating, depending on the partition coefficients. The analytes may then be desorbed by heat in the injection port of a gas chromatograph. Although the specific derivatization mechanism mentioned above has the advantage of taking place in the aqueous phase, a substantial percentage of organic solvents are still present after derivatization is completed. Limitations of the commercial fibers include low durability when working at high temperature, low tolerance
to extreme pH and instability in some organic solvents.8–11 Some authors, in order to preserve the lifetime of the fiber when derivatization is a necessity or the matrix is harsh, have employed headspace sampling.10,12
Recently, investigations into more stable and selective coatings have begun13,14 and fibers have been prepared utilizing the sol–gel process to give enhanced stability to extreme temperatures and exposure to solvents.15 These enhanced stabilities are attributed to the polymeric coating of the SPME fiber being chemically bonded to the silica substrate instead of attached by physical deposition. The sol–gel process consists of acid-catalyzed hydrolysis of the alkoxide groups of a silica precursor followed by condensation of the resulting silanol. In preparing coatings on silica substrates for SPME, condensation reactions of the silanol groups on the fused silica fiber with those of the silica precursors provide chemical anchorage of the sol–gel network to the surface of the silica fiber. Thus, a stable surface-bonded polymeric coating is formed. The main objective of this work is to illustrate the capabilities
of laboratory-prepared SPME fibers based on sol–gel technology for the analysis of seleno amino acids using ICP-MS detection.
Experimental
Instrumentation
An Agilent 6890 GC directly coupled to an Agilent 7500 ICP-MS with a heated transfer line was employed. Power supply and temperature control for the heated transfer line were managed through the ChemStation software. Instrument operating conditions are shown in Table 1. Selenium isotopes, 77Se, 78Se and 82Se, were monitored. A Shimadzu GC-17A GC equipped with FID detection was also used.
| Injector temperature |
310 °C (splitless mode) |
| Oven temperature program |
50 °C for 10 min; 20 °C min−1 to 210 °C |
| GC column |
HP-5, 30 m length, 0.32 mm id, 0.25 μm film thickness |
| GC mobile phase |
Helium 1 mL min−1 |
| Transfer line |
250 °C to torch, silica capillary |
| Plasma rf power |
1300 W |
| Sampling depth |
6 mm (1 mm orifice Pt cones) |
|
ICP-MS gas flows
|
| Plasma |
14 L min−1 |
| Auxillary |
1 L min−1 |
| Carrier |
1.6 L min−1 |
| Isotopes monitored |
77Se, 78Se and 82Se |
Reagents and standards
Standards.
Se-methionine, Se-ethionine and Se-cystine standards were obtained from Sigma (Milwaukee, WI, USA). Working standards were prepared fresh by serial dilution of stock standards.
SPME fiber coating preparation
The procedure used was a modification of that published by Chong et al.15 The sol–gel solution was prepared by mixing the following reagents: 150 μL of methyltrimethoxysilane, 100 μL of trifluoroacetic acid and 90 mg of hydroxy-terminated polydimethylsiloxane. The reagents were mixed for 20 min using a magnetic stirrer and stir plate and the resulting solution was used for coating the fiber. The silica fiber was dipped into the sol–gel solution five times for 10 min each time. The PDMS polymer coating was end-capped by dipping the fiber into a solution of trimethylmethoxysilane + methanol (4∶1 v/v) for 2 min. Finally the fiber was conditioned in a GC injection port at 250 °C for 2 h.
Silica fiber substrate
Small diameter fibers lacked the mechanical strength to withstand the extension and retraction necessary for typical fiber operation, but larger diameter fibers destroyed the GC septum after a few injections. Best results were achieved with SPME fibers purchased from Supelco (Bellefonte, PA, USA) and this allowed the use of a commercial SPME syringe. Fibers whose coatings had previously been exposed to aqueous samples containing the derivatizing solvents were used. The remaining coating was completely removed from the silica by dipping the fiber tip into concentrated sulfuric acid. The silica tip was then soaked in 1 M KOH solution so that all surface groups were in the form of Si-O-H to better effect the sol–gel reaction.16
Derivatization/extraction into chloroform for direct injection
A 50 μL volume of an alcohol + pyridine mixture (3∶1 v/v) and 10 μL of alkylchloroformate were added to a 100 μL volume of aqueous sample. This mixture was shaken for 2 min. 500 μL of chloroform were added and the sample was again shaken for 2 min. A 2 μL sample of the chloroform extract was injected into the gas chromatograph.
A 1200 μL volume of an alcohol + pyridine mixture (3∶1 v/v) was added to 2400 μL of aqueous sample. This mixture was magnetically stirred for 10 min. 250 μL of alkylchloroformate were added and the sample was shaken for 2 min with venting.
SPME
A Teflon-coated magnetic stirrer was added to a glass vial with a PTFE (Teflon)-coated septum containing the derivatized sample and the vial was placed on a magnetic stir plate. The SPME fiber was exposed to this derivatized aqueous sample for an optimally chosen time of 5 min.
Results and discussion
The use of SPME in the analysis of the selenium analogs of amino acids represents an interesting area of research, especially in clinical chemistry.17 The use of Se-selective detectors such as rf-GD-MS or ICP-MS provides exceptional limits of detection that can be further improved with the use of preconcentration techniques such as SPME. However, the number of coating materials is limited and, in some cases, not adequate for the analytes under study. Selecting fiber coatings with strong affinities for the analytes enhances preconcentration and provides better sensitivity. Supelco recommends the 100 μm PDMS fiber for low molecular weight compounds while the 7 μm PDMS fiber is suggested for larger molecular weight or semi-volatile compounds, because, generally, thin coatings are most effective for absorbing/desorbing semi-volatile analytes.
In this study, both fibers were initially evaluated for the selective extraction of the Se-methionine derivative. In addition, a relatively new commercial fiber (Carboxen/PDMS) was also tested for the preconcentration of the analyte when sampling the headspace and using rf-GD-MS as a selective Se detector. However, the low volatility of the derivatized Se-methionine provided no results with any of the three fibers tested in this way. Attempts to heat the solution in order to force the absorption of the derivatized Se-amino acid were also unsuccessful, although the pyridine (used as the reaction catalyst) was vaporized and absorbed on the fiber. Therefore, immersion of the fibers in aqueous solution was necessary.
In sampling by immersion, the Carboxen/PDMS fiber proved to be the most effective to preconcentrate the Se-methionine derivative followed by the100 μm PDMS fiber and then by the laboratory-made sol–gel PDMS fiber. The analyte showed no affinity for the 7 μm PDMS fiber. However, the chromatogram obtained using the Carboxen/PDMS fiber exhibited desorption problems (tailing) as can be observed in Fig. 1B. The first peak observed in both chromatograms (Fig. 1) is due to pyridine, which is used as the reaction catalyst. This raised concerns regarding the extraction by SPME fibers of large amounts of derivatizing solvents that remained in the aqueous sample, which could then minimize the extraction effectiveness for the analytes. Furthermore, other authors have noted that the Carboxen/PDMS fiber absorbs larger amounts of extraneous material in the sample when compared to the PDMS fiber. They hypothesized that this was
due to the adsorption capacity of the Carboxen/PDMS fiber as compared with the absorption capacity of the PDMS fiber.18 This also may help to explain the progressive deterioration of the Carboxen/PDMS coating that occurred after several extractions of the derivatized samples. Because of these limitations and because of the temperature restrictions of the commercial PDMS fibers, the sol–gel laboratory-made fibers were investigated for the purpose of assessing if adequate sensitivity was provided with the enhanced durability.
 |
| | Fig. 1
Extracted ion chromatograms of Se-methionine ethyl derivative, monitoring m/z 80 with rf-GD-MS. Chromatogram A was obtained with a 100 μm PDMS fiber; chromatogram B was obtained with a 75 μm Carboxen/PDMS fiber.
| |
Characterization of the sol–gel PDMS fiber
To gain a better understanding of the extraction by SPME fibers, further work was conducted using a more universal detector, namely flame ionization. Several parameters were initially optimized in order to obtain maximum recoveries for the Se-methionine derivative: derivatizing reagent, polymer coating length, solvent effect, extraction/desorption time and pH adjustment/salt addition.
Alkyl group of the derivatization reagent
Other researchers have found that the isobutyl derivative, as compared to the ethyl, allyl and butyl derivatives, was most sensitive to extraction by a PDMS-coated fiber when analyzing for the sulfur analogs of the seleno amino acids.17 This was again found to hold true in the derivatization and solid-phase microextraction of amines. The PDMS fiber gave a factor of two better sensitivity for extraction with the isobutyl derivative in comparison with the ethyl derivative.19 Although initial experiments were performed with the ethyl derivative (with rf-GD-MS detection), in order to increase the absorption of the seleno amino acid derivatives with the sol–gel PDMS coating, subsequent work for this paper was performed using isobutanol and isobutylchloroformate for the seleno amino acid derivatizations.
Effect of polymer size
The size effect of the PDMS polymer used in the sol–gel process was examined. Molecular weights of 570 (n = 8), 3400 (n = 46), and 17000 (n = 230) g mol−1 were used. Fig. 2 shows that, as the length of the polymer chain increased, the extraction efficiency of the polymer-coated fiber decreased. This might be explained by the greater viscosity encountered with increasing molecular weight. This could inhibit mobility in the sol–gel solution resulting in poor coverage of the silica fiber. Another possibility is that the greater mobility of the smaller molecular weight polymer allowed for attachment to the silica surface followed by subsequent polymerization of PDMS (n = 8) units. However, these theories remain as speculation because further characterization was not done, i.e., the exact thickness of the three coatings was not determined. These experiments were performed solely to optimize
the size of the PDMS precursor, but further work is being conducted in order to fully characterize the fiber surface.
GC-FID
The use of a universal detector such as FID allowed observation of several unexpected effects on the absorption of the Se-methionine. Although the sol–gel fiber withstood the organic solvents of the derivatization procedure without being compromised, the chromatograms obtained from the analysis of an aqueous sample of derivatized selenomethionine showed substantial solvent peaks, as can be seen in Fig. 3. In addition, at this point, a quantitative analysis showed that the fiber did not produce the expected preconcentration of the analyte. Because the PDMS-coated fiber extracts under the principle of absorption (non-competitive) rather than adsorption (competitive), it was assumed at this point that the large solvent concentration in the aqueous sample increased the solubility of the derivatized seleno amino acids within the aqueous sample. This caused a decrease in the partition coefficient of the analyte and subsequently the extraction amount was
reduced. Other authors have noted similar phenomenon. Lee et al.20 observed an inhibition in the extraction of acidic herbicides in waters with a high humic content. Urruty and Montury21 studied the influence of ethanol concentrations in the analysis of pesticides and concluded that, by increasing ethanol concentrations, variations in the partition coefficients of the organic analytes were induced. Finally, Wu and Huang22 observed a diminished extraction when the methanol concentration of the aqueous sample increased, and concluded that the decreased polarity of the sample caused a decrease in the partition coefficient.
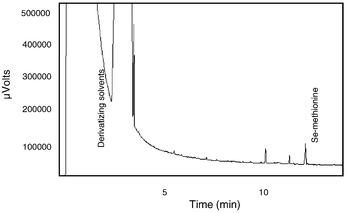 |
| | Fig. 3
Chromatogram of Se-methionine utilizing FID detection; extraction performed with sol–gel-prepared PDMS fiber.
| |
Therefore, the derivatization/solid-phase microextraction procedure was modified to include an additional step. After the reaction was complete, the derivatizing solvents were removed by allowing the samples to evaporate under a gentle stream of nitrogen. The time of evaporation was closely controlled and sample volume was adjusted, if necessary, once the evaporation process was complete. As a result, although the solvent peaks did not disappear completely from the chromatograms, the sensitivity was increased as the concentration effect was observed. It was consequently concluded that, as a result of removing the solvents from the aqueous phase, the solubility of the hydrophobic analytes decreased and therefore more readily partitioned into the SPME fiber coating. Table 2 lists a concentration effect of 7.2 when SPME was used. Most other researchers have claimed a minimal preconcentration effect of 10 and this lower value of 7.2 might be attributed to
the residual derivatizing solvents in the aqueous sample. Also to be noted in Table 2 is the greater sensitivity of the SPME method when operated with splitless injection. Because SPME is a solventless technique, i.e., analytes are extracted into a polymeric coating rather than an organic solvent, it allows the operation of this mode of injection and, subsequently, can offer even greater sensitivities than conventional liquid sample introduction.
Table 2
Comparison of the response of the isobutyl derivative of seleno methionine for chloroform extraction vs. SPME
| Extraction type |
Injection mode |
Normalized area |
| SPME |
Splitless |
190 |
| SPME |
Split |
7.2 |
| Chloroform extraction |
Split |
1.0 |
Optimization of absorption: extraction time profile
Solid-phase microextraction depends on the establishment of an equilibrium between the analytes in the aqueous phase and the fiber coating. One of the first steps in the development of a SPME method is the determination of this equilibration time. It is not an exhaustive extraction, so different extraction times must be evaluated to determine the time at which equilibrium is attained. Extraction of the seleno amino acid derivatives for various lengths of time yielded the results displayed in Fig. 4. The optimal extraction time was found to be at 5 min. After this time, the amount of seleno amino acids within the fiber coating began to decrease. Considering again the absorptive extraction mechanism of the PDMS coating, it is possible that this phenomenon is a result of modification of the coating characteristics due to the large amount of solvent present in the coating after only a few minutes. Similarly, Aguerre et al.23
noted the competitive extractions between the analytes and other organic substances and published extraction time profiles that showed a decrease in the extraction amount over time. Gorecki et al.24 circumvented a similar problem by shortening the extraction time, thus limiting the uptake of the large number of molecules of the interfering species. Therefore, an extraction time of 5 min was chosen for these analytes as optimum.
 |
| | Fig. 4
Extraction time profile for seleno methionine.
| |
Optimization of desorption time
As in any extraction procedure, it is imperative to eliminate any traces of carryover that may detrimentally affect the next sample analysis. It is usual in analysis using SPME to evaluate the time and temperature of the desorption process in order to eliminate this potential problem. Unfortunately, the boiling points for the seleno amino acid derivatives are not cataloged in the usual chemical resources. Boiling points of the derivatives were estimated by Beilstein software using the Joback fragmentation method and were found to be approximately 300 °C. Although the PDMS commercial fibers cannot withstand this operating temperature, due to problems with bleed,9 sol–gel-prepared SPME fibers perform well at these high temperatures. Therefore, the injection port temperature of the GC was maintained at 310 °C and the desorption time was set at 5 min. Examination of fiber blanks showed no carryover under these conditions.
Addition of salt and acid
In the extraction of organic compounds from aqueous samples, acids, bases and salts are frequently employed to improve extraction efficiencies. By altering the pH and increasing the ionic strength of the aqueous sample, the affinity of the organic compounds for the aqueous phase is decreased and extraction efficiency is subsequently increased. In this application, it was necessary to exercise care with the addition of acid; pH values of less than 2 would promote the hydrolysis of the newly esterified seleno amino acids. Results of these experiments are shown in Table 3. Optimal extraction efficiency was obtained with the adjustment of the pH to 3.0 and the addition of 10% NaCl.
Table 3
Effect of pH adjustment and salt addition on the extraction efficiency of the sol–gel-prepared PDMS fiber
| |
Normalized area |
%RSD |
| Aqueous sample only |
1.0 |
5.5 |
| Aqueous sample/HCl |
2.2 |
2.8 |
| Aqueous sample/Na2SO4 |
1.4 |
NA |
| Aqueous sample/HCl/Na2SO4 |
3.8 |
8.9 |
Analytical performance characteristics
Under the optimized extraction conditions, the analytical performance characteristics were evaluated using ICP-MS as a selective Se detector. An extracted ion chromatogram for m/z 77 is shown in Fig. 5. Figures of merit are shown in Table 4. A very low limit of detection was obtained for all the compounds (in the ppt range as Se-amino acids derivatives). However, reproducibility was not as good as that typically obtained for SPME with commercial fibers. This is probably due to irregularities present in the sol–gel film; films deposited in a commercial production process would be much more consistent than those prepared in our laboratory. Additionally, the added step of a derivatization reaction may be a contributing factor. Further studies will be done to find a more homogeneous coating procedure than described above. This should improve the reproducibility. However, the fiber developed showed good
results in terms of durability and sensitivity. Linearity seemed to be reasonably good up to 1 ppm, which was the range studied.
Table 4
Figures of merit for the analysis of seleno amino acids by SPME-GC-ICP-MS
| |
Se-methionine |
Se-ethionine |
Se-cystine |
|
R
2
|
0.963 |
0.987 |
0.999 |
| LOD (ppt) as seleno amino acid derivative |
16 |
14 |
29 |
| RSD of 100 ppt sample (n = 5) |
23% |
17% |
22% |
Conclusions
Sol–gel-prepared PDMS fibers for use in solid-phase microextraction show potential as better alternatives to the non-chemically bonded commercial fibers in this particular application. Chemically bonding the PDMS polymer to the silica substrate provides a more rugged fiber, which is less susceptible to the breakdown caused by the organic components in the derivatization procedure. Modifications made to the derivatizing reagents, adjustment of the pH and the addition of salt, allowed the PDMS sol–gel fiber to compete favorably in terms of sensitivity, while providing the necessary enhanced durability. Better reproducibility will be attained as the coating process for the sol–gel-based fibers is improved.
Acknowledgements
The authors would like to thank Agilent Technologies for the use of a Model 7500s ICP-MS and a Model 6890 GC. We would also like to acknowledge the National Institutes of Environmental Health Sciences for grant # ESO4908, project 5 and project 8, for partial funding of this research.
References
- K. Pyrzynska, Analyst, 1996, 121, 77–83 RSC.
- S. M. Bird, H. Ge, P. C. Uden, J. F. Tyson, E. Block and E. Denoyer, J. Chromatogr., A, 1997, 789, 349–359 CrossRef CAS.
-
K. L. Ackley, K. L. Sutton and J.
A. Caruso, Elemental Speciation—New Approaches for Trace Element Analysis, Elsevier, New York, 2000 Search PubMed.
- M. Vazquez Pelaez, M. Montes Bayon, J. I. Garcia Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 2000, 15, 1–6 RSC.
- P. Husek, J. Chromatogr., B, 1998, 717, 57–91 CrossRef CAS.
- L. Pan and J. Pawliszyn, Anal. Chem., 1997, 69, 196–205 CrossRef CAS.
- Z. Zhang, M. J. Yang and J. Pawliszyn, J. Anal. Chem., 1994, 66, 844A–853A Search PubMed.
- R. C. B. Aranda, J. Chromatogr., A, 1998, 829, 401–406 CrossRef CAS.
- M. L. Bao, F. Pantani, O. Griffini, D. Burrini, D. Santianni and K. Bargieri, J. Chromatogr., A, 1998, 809, 75–87 CrossRef CAS.
- H. L. Lord and J. Pawliszyn, Anal. Chem., 1997, 69, 3899–3906 CrossRef CAS.
- A. A. Boyd-Boland and J. Pawliszyn, Anal. Chem., 1996, 68, 1521–1529 CrossRef CAS.
- T. Gorecki and J. Pawliszyn, Anal. Chem., 1996, 68, 3008–3014 CrossRef CAS.
- M. Ligor, M. Scibiorek and B. Buszewski, J. Microcolumn Sep., 1999, 11, 377–383 CrossRef CAS.
- M. E. Cisper, W. L. Earl, N. S. Nogar and P. H. Hemberger, Anal. Chem., 1994, 66, 1897–1901 CrossRef CAS.
- S. L. Chong, D. W, James D. Hayes, B. W. Wilhite and A. Malik, Anal. Chem., 1997, 69, 3889–3898 CrossRef CAS.
- Y. Guo, G. A. Imahori and L. A. Colon, J. Chromatogr., A, 1996, 744, 17–29 CrossRef CAS.
- S. W. Myung, M. Kim, H. K. Min, E. A. Yoo. and K. R. Kim, J. Chromatogr., B, 1999, 727, 1–8 CrossRef CAS.
- Z. Mester, R. E. Sturgeon and J. W. Lam, J. Anal. At. Spectrom., 2000, 15, 1461–1465 RSC.
- L. Pan, J. M. Chong and J. Pawliszyn, J. Chromatogr., A, 1997, 773, 249–260 CrossRef CAS.
- M. R. Lee, R. J. Lee, Y. W. Lin, C. M. Chen and B. H. Hwang, Anal. Chem., 1998, 70, 1963–1968 CrossRef CAS.
- L. Urruty and M. Montury, J. Agric. Food Chem., 1996, 44, 3871–3877 CrossRef CAS.
- Y. C. Wu and S.
D. Huang, Anal. Chem., 1999, 71, 310–318 CrossRef CAS.
- S. Aguerre, C. Bancon-Montigny, G. Lespes and M. Potin-Gautier, Analyst, 2000, 125, 263–268 RSC.
- T. Gorecki, P. Martos and J. Pawliszyn, Anal. Chem., 1998, 70, 19–27 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here. 
