Immunofiltration as sample cleanup for the immunochemical detection of β-agonists in urine
Willem
Haasnoot
*a,
Anniek
Kemmers-Voncken
a and
Danny
Samson
b
aState Institute for Quality Control of Agricultural Products (RIKILT), P.O. Box 230, 6700 AE Wageningen, The Netherlands. E-mail: w.haasnoot@rikilt.wag-ur.nl; Fax: +31 317 417717; Tel: +31 317 475603
bLaboratory of the National Inspection Service for Meat, Livestock and Animal Products (L-RVV), P.O. Box 144, 6700 AC Wageningen, The Netherlands
First published on 13th December 2001
Abstract
Despite the ban of the European Union on use of drugs to improve animal growth, occasionally β-agonist drugs are still found in samples from cattle. Over time, the specified limits for the detection of these illegal drugs have been lowered. To improve the immunochemical screening of urine samples to detect lower levels of several β-agonists, immunofiltration (IF) was applied for sample cleanup in combination with a β-agonist-ELISA. In the applied IF format, free (non-immobilised) anti-salbutamol polyclonal antibodies were mixed with the urine sample in an ultra-filtration device (cut off 30 kDa) and the sample was removed by centrifugation. The antibody bound β-agonists were freed from the antibodies by the addition of a mixture of methanol and 0.1 M acetic acid (1∶1; v/v) and centrifugation. The filtrate, containing the free β-agonists, was evaporated to dryness and the residue dissolved in buffer, an aliquot of which was analysed with the β-agonist ELISA. Compared with the direct β-agonist ELISA, this IF cleanup procedure resulted in a 30-times lower limit of detection (LOD) of 0.14 ng ml−1 (salbutamol equivalents). The anti-salbutamol antibodies recognised several β-agonists and the combination of IF with the β-agonist ELISA was found suitable for the detection of at least ten β-agonists in urine with comparable LODs.
Introduction
Several investigations have demonstrated that the use of β-agonist drugs in food-producing ruminants (cattle, calves, pigs and sheep) have a positive effect on growth as well as on the protein/fat tissue ratio. Within the European Union, the use of any drug to improve animal growth is banned.1 Despite this ban, β-agonist drugs were frequently found in samples from cattle.2 In order to control misuse in The Netherlands, different sample materials (urine, faeces, cattle feed, tissues, hair, eyes, etc.) are taken for analysis for the presence of β-agonists. Of these samples, urine, which can be sampled either at farms or in slaughterhouses, is still most frequently analysed. For the detection of β-agonists in urine, a two-stage control programme using microtitre plate enzyme linked immunosorbent assays (ELISAs) for screening3 and GC-MS for confirmation4,5 is applied. For a first screening at the farm, an on-site test, based on an ELISA in polystyrene tubes, was developed.6 In this on-site test and in the microtitre plate β-agonist ELISA, a mixture of antibodies raised against clenbuterol and salbutamol was used, which made these tests sensitive to a range of β-agonists. The performances of these tests was compared previously.6 In the on-site test, urine samples were analysed without a sample preparation and, in the microtitre plate ELISA, samples were analysed after a five-fold dilution in buffer. The absence of a sample preparation resulted in varying background responses with urine samples from different animal sources, which had a negative effect on the limits of detection (LODs). For example, with the on-site test, only incurred bovine urine samples with a level above 3 ng of clenbuterol per ml were found positive, while in calf urine, due to lower matrix effects, an LOD of 1 ng ml−1 could be achieved.6The application of sample preparation prior to an immunoassay could reduce matrix interferences and gave the possibility for concentrating the analytes of interest, both having a positive effect on the LOD of the assay. For instance, for the detection of clenbuterol with a biosensor immunoassay, urine samples were extracted with tert-butyl methyl ether (TBME) and eight times concentrated, which resulted in an LOD of 0.27 ng ml−1.7 However, TBME is not suitable for the extraction of β-agonists such as salbutamol and terbutaline.7,8 A fast and simple extraction on Empore membranes with strong cation exchange properties was described by Vanoosthuyze et al.,9 but the recoveries for salbutamol and terbutaline were also low (approx. 10%).
The best efficiency for the extraction of several β-agonists, including salbutamol and terbutaline, was obtained with isobutanol (2-methylpropan-1-ol) and, in combination with the β-agonist ELISA, LODs below 1 ng ml−1 were obtained.3 However, hydroxylated β-agonists (as for instance salbutamol) are excreted in urine mainly as glucuronide and/or sulfate derivatives10 and, prior to an extraction procedure, a time consuming hydrolysis (i.e., with Helix pomatia juice) had to be applied.
Hellenäs et al.11 used immunofiltration (IF) for preparation of urine samples for detection of clenbuterol with a biosensor immunoassay. The antibodies were mixed with the urine sample in an ultra-filtration (UF) device (cut off 30 kDa) and, after a short incubation and centrifugation, the filter was washed with buffer. The antibodies with bound clenbuterol were detected in the biosensor immunoassay with clenbuterol immobilised on the sensor surface. In urine, an LOD of 0.9 ng ml−1 was obtained. The application of IF, as a sample pre-treatment for clenbuterol in urine, in combination with a clenbuterol ELISA, has also been described.12 In this IF format, the bound clenbuterol was freed from the antibodies by the addition of 0.3 ml of 0.1 M acetic acid and centrifugation. The pH of the eluted fraction was neutralised by the addition of strong buffer and aliquots were tested in the clenbuterol ELISA. Using this procedure, matrix inferences of urine samples in the ELISA were reduced substantially, which resulted in a low LOD of 0.05 ng ml−1.
In the present study, we aimed for a broad immunochemical screening assay with low LODs for different β-agonists in urine and used antibodies raised against salbutamol for the sample cleanup by IF, in combination with the β-agonist ELISA . The assay was tested for its performance using: (i) standard solutions of different β-agonists, (ii) blank bovine urine samples (with and without the addition of salbutamol), (iii) incurred urine samples with low concentrations of different β-agonists, with the IF-ELISA results being compared with those obtained by GC-MS and (iv) urine samples which gave false positive results in the ELISA following sample preparation by the isobutanol extraction.
Experimental
Materials
The production of the polyclonal antisera, raised in rabbits against clenbuterol– and salbutamol–bovine serum albumin, was described previously.3 Microcon® centrifugal filter devices (YM-30) were obtained from Millipore Corporation (Bedford, MA, USA).Clenbuterol hydrochloride, fenoterol hydrobromide, metaproterenol hemisulfate, isoxsuprine hydrochloride and antifoam A emulsion were obtained from Sigma (St. Louis, MO, USA). Salbutamol sulfate and terbutaline sulfate were obtained from Bufa-Chemie (Castricum, The Netherlands). Dr. M. W. F. Nielen (RIKILT, Wageningen, The Netherlands) supplied us with standards of mabuterol, mapenterol, bromobuterol, cimbuterol, tulobuterol, carbuterol, pirbuterol, ractopamine, clenpenterol, cimaterol, clenproperol and clencyclohexerol, which were gifts from different sources.
Microtitre plates (96-wells) were obtained from Greiner (Frickenhausen, Germany) and solutions of tetramethylbenzidine (TMB) peroxidase substrate and peroxide were supplied by Kirkegaard and Perry Labs (Gaithersburg, MD, USA). Goat anti-rabbit antibody was supplied by Caltag Laboratories Inc. (Burlingame, CA, USA).
Equipment
A Wellwash Model 4-MK2 microplate washer (Denley Instruments, Billinghurst, UK), an Argus 400 microplate reader (Canberra Packard, Downers Grove, IL, USA) and a Sigma Model 302k centrifuge (Osterode, Germany), using an angle rotor suitable for 24 reaction-vials (e.g. Eppendorf), were used. The Reacti-Therm™ III heating module was from Pierce (Rockford, IL, USA).The GC-MS system (Hewlett Packard, Rockville, MD, USA) consisted of a Model 5890 gas chromatograph, a Model 7673 auto sampler and a Model 5970 mass-selective detector. A DB-5 column (30 m × 0.25 mm id) (J&W Scientific, Folsom, CA, USA) with a film thickness of 0.25 μm was used with helium as the carrier gas.
Immunofiltration procedure
To the UF-device [Microcon® centrifugal filter devices (YM-30)], 0.25 ml of urine (pH adjusted to 7 ± 1) was added, followed by 0.25 ml of antiserum (anti-salbutamol) which was 50 fold diluted in PBS (5.4 mM sodium phosphate 1.3 mM potassium phosphate 150 mM sodium chloride; pH 7.4). After mixing on a vortex, the UF-device was centrifuged at 5000g for 15 min. To wash away the remaining sample, 0.2 ml of PBS was added to the retentate and the device was centrifuged again for 15 min. The filtrate was removed and to free the antibody-bound β-agonists, 0.3 ml of a mixture of methanol and 0.1 M acetic acid (1:1; v/v) was added to the retentate. After centrifugation (45 min at 5000g), the filtrate was evaporated at 50 °C under a stream of nitrogen. The sample extract was dissolved in 0.25 ml PBS (equivalent to 1 ml of sample per ml) and 50 μl portions were transferred to the β-agonist ELISA.Isobutanol extraction procedure
The pH of a 1 ml sample of urine was adjusted to 4.8 ± 0.2 by adding a few drops of 1 M acetic acid. Afterwards, Helix pomatia juice (25 μl) was added and the mixture was incubated for 2 h at 55 °C, or overnight at 37 °C. The pH of the hydrolysed urine was adjusted to 9.5 ± 0.5 by adding a few drops of 0.1 M sodium hydroxide, and 2 ml of isobutanol was added. After vortex mixing for 1 min and centrifugation for 10 min at 1500g, 1 ml of the isobutanol was evaporated at 50 °C under a stream of nitrogen. The residue was dissolved in 0.5 ml of PBS (equivalent to 1 ml of sample per ml) and 50 μl portions were transferred to the β-agonist ELISA.β-Agonist ELISA
Microtitre plates were coated overnight with 100 μl aliquots of goat anti-rabbit IgG (5 μg ml−1 in 50 mM sodium carbonate; pH 9.6) at 4 °C. Plates were washed three times with washing buffer (PBS, containing 0.05% Tween-20 and 0.004% antifoam). Aliquots of 50 μl of standard solutions of salbutamol (0.05–5 ng ml−1 in PBS) or 50 μl of sample were added to the wells followed by 25 μl of salbutamol–HRP diluted in PBS (1∶30000; v/v) and 25 μl of the antibody mixture (anti-salbutamol–anti-clenbuterol; 15∶1; v/v) diluted in PBS (1∶1000; v/v). The plate was incubated for at least 2 h at 4 °C and, after washing three times with washing buffer, the quantity of bound peroxidase was determined by adding 100 μl of a tetramethylbenzidine (TMB) peroxidase substrate. After incubation in the dark for 20–30 min at room temperature, the reaction was stopped by adding 100 μl of 1 M phosphoric acid and the coloured product was measured at 450 nm.Confirmation method based on GC-MS
The identification and quantification of the β-agonists present in the urine samples was performed by GC-MS according to a standard operating procedure applied at L-RVV (Wageningen, The Netherlands).In short, the applied procedure was as follows. A mixture of internal standards (deuterated β-agonists) was added to the urine sample (1 ml) and, after the addition of water (4 ml), the pH was adjusted to 4.8 ± 0.2 with acetic acid. Helix pomatia juice (25 μl) was added and the mixture was incubated for 2 h at 55 °C or overnight at 37 °C. After the addition of 5 ml of a phosphate buffer, the pH was adjusted to 7.4 ± 0.2 by means of an acetic acid solution or a sodium hydroxide solution. The mixture was transferred to an immunoaffinity column (containing 1 ml of CNBr-activated Sepharose with immobilised anti-clenbuterol) and thereafter, the column was washed with water. The bound β-agonists were eluted using 5 ml of a mixture of methanol and 0.1 M acetic acid (7∶3; v/v) and the eluted fraction was concentrated until 1 ml by evaporation. After the addition of a 1 M phosphate buffer, the pH was adjusted to 7.6 ± 0.2 by means of an acetic acid solution or a sodium hydroxide solution and the mixture was transferred to an activated C18 solid phase column (1 ml; Mallinckrodt Baker, Deventer, The Netherlands). Thereafter, the column was washed with water and dried under a stream of nitrogen. The bound β-agonists were eluted by means of a mixture of methanol and acetonitrile (85∶15; v/v) and the eluted fraction was evaporated to dryness at 50 °C under a stream of nitrogen. After derivatisation at 70 °C for 30 min, using a mixture of N,O-bis (trimethylsilyl)trifluoroacetamide (BSTFA) and ethyl acetate (1∶1; v/v), the sample was transferred to a GC-MS vial and the solvent was evaporated at 35 °C under a stream of nitrogen. The residue was dissolved in ethyl acetate (10 μl) of which 3 μl was injected in the splitless mode into the GC-MS system. The analytes were considered to have been positively identified if the GC retention times of the derivatives and the ratios of the intensities of the fragments agreed with those of the standards to within ±5 s and ±10%, respectively. The concentration of a β-agonist in the sample was quantified using the intensity of a relevant fragment of the respective internal standard with a known concentration.
Samples
Twenty lyophilised reference blank bovine urine samples (code BOV01-BOV20) were obtained from the bank of reference samples prepared by the European Community Reference Laboratory (CRL) the National Institute of Public Health and Environmental Protection (RIVM, Bilthoven, The Netherlands).13 These samples were taken from a broad range of animals including veal calves, fattening bulls, heifers, pregnant cows and mature bulls. Each sample was reconstituted in 5 ml of deionised water.Other negative and positive urine samples were selected from those taken by the Dutch National Inspection Service for Meat, Livestock and Animal Products (RVV) in connection with the National Programme. The samples were stored at −20 °C until used.
Results and discussion
Optimum conditions for IF
Under the conditions described in the experimental section, the calibration curve for salbutamol in the β-agonist ELISA showed a measurement range from 0.05 to 5 ng ml−1 with 0.25 ng ml−1 at 50% inhibition (see Fig. 1). In IF, the amount of antibodies added to the UF-device determines the maximum amount of analyte binding. Using IF in combination with the ELISA, an amount of antibodies which bind a maximum concentration of 5 ng ml−1 of a β-agonist should be enough. To find the optimum amount of antibodies, volumes of 0.25 ml of the anti-salbutamol raw antiserum (10, 50 and 250 times diluted) were mixed with 0.25 ml of a standard solution of salbutamol (5 ng ml−1; 1.25 ng absolute) in the UF-device. After centrifugation, the concentrations of salbutamol in the filtrate (non-bound salbutamol) were measured by ELISA, which indicated the percentages of antibody-bound salbutamol in the retentate. From this experiment, 0.25 ml of a 50 times diluted antiserum (equal to 5 μl of the raw serum), which bound approx. 85% of the added salbutamol, was chosen for further experiments. In former experiments with anti-clenbuterol,12 1 μl of the raw serum sufficed to bind 90% of a comparable amount of clenbuterol. The need to use less anti-clenbuterol, compared to anti-salbutamol, in IF could be explained by the higher amount of specific antibodies in this antiserum (final dilution of 1∶10000 in the ELISA) compared to the anti-salbutamol serum (final dilution of 1∶1000 in the ELISA).3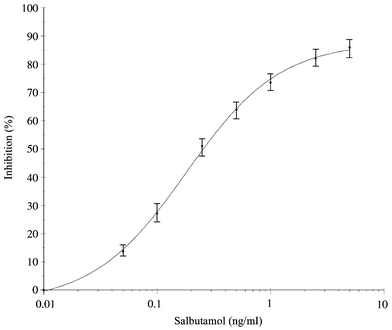 | ||
| Fig. 1 Salbutamol calibration curve obtained in the β-agonist ELISA (an average of seven curves obtained on four different days). | ||
In order to elute the bound salbutamol from the antibodies, different volumes of several solvents (mixtures of methanol with buffer and 0.1 M acetic acid) were tested. The best results were obtained with 0.3 ml of a mixture of methanol with 0.1 M acetic acid (1∶1; v/v) which resulted in a recovery of approx. 85%. This elution solvent could easily be evaporated to dryness and, after dissolving the residue in PBS, the pH of the solution was neutral and suitable for transfer to the β-agonist ELISA.
LOD of the IF-ELISA
To prove that IF had a positive effect on the reduction of matrix effects, the 20 reference blank bovine urine samples were analysed with the direct β-agonist ELISA (undiluted and five-fold diluted samples) and after IF. As shown in Fig. 2, the undiluted urine samples gave high background responses and a high variation (73 ± 25% inhibition overall), which made assay of undiluted samples unsuitable. A five-fold dilution of the urine samples in buffer reduced the matrix interferences and lower background responses were obtained. The average background response (43 ± 21% inhibition) corresponded with 1.4 ± 1.1 ng ml−1 (salbutamol equivalents), which resulted in an LOD of 4.7 ng ml−1 (average + 3 times standard deviation, s). The average background response after IF showed 18 ± 8% inhibition, which corresponded with 0.065 ± 0.026 ng ml−1 (salbutamol equivalents) and with an LOD of 0.14 ng ml−1. Therefore, compared with the direct assay using five-fold diluted urine samples, the application of IF-purified and undiluted urine samples resulted in a 30 times lower LOD in the ELISA. Possible explanations for this reduction in matrix interference are: (i) the use of 50 times more antibody in IF compared to the amount in an ELISA-well and (ii) the several interactions involved in the competitive direct ELISA format (anti-salbutamol to goat anti-rabbit; anti-salbutamol to salbutamol–HRP and anti-salbutamol to the free β-agonists) which might be influenced by the urine matrix while, in IF, the only interaction is between the relatively high amount of anti-salbutamol and the free β-agonists. | ||
| Fig. 2 Percentages of inhibition obtained in the β-agonist ELISA with the 20 reference blank bovine urine samples using a direct immunoassay (no sample preparation), with undiluted and five-fold diluted samples, and after IF. | ||
Salbutamol recovery with IF-ELISA
The 20 reference blank bovine urine samples were also analysed after the addition of salbutamol at the 1 and 5 ng ml−1 level. As shown in Fig. 3, the difference between the average inhibition obtained for the blank samples and the spiked samples was greater with IF than with the direct ELISA (undiluted or five-fold diluted) and the variation was less. With IF, the average recoveries of salbutamol added to urine samples at the 1 and 5 ng ml−1 levels, were 77 ± 19% and 58 ± 16%, respectively. Due to the lower recoveries at the higher levels and a flattening calibration curve above a concentration of 1 ng ml−1 (see Fig. 1), the quantitative results above the 1 ng ml−1 level were not accurate. However, for a screening assay with an LOD far below this level, this was not a problem.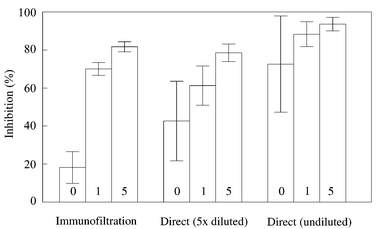 | ||
| Fig. 3 Average percentages of inhibition obtained in the β-agonist ELISA with the 20 reference blank bovine urine samples and the same samples spiked with salbutamol at 1 and 5 ng ml−1. The samples were analysed directly (undiluted and five-fold diluted) and after IF. | ||
IF-ELISA for other β-agonists
To prove that the combination of IF and the β-agonist ELISA worked for different β-agonists, standard solutions of the β-agonists at concentrations of 1 and 5 ng ml−1 were analysed with the ELISA directly and after IF (see Table 1). At the 1 ng ml−1 level, eleven of the β-agonists tested caused significant inhibition (>40%) after IF. Compared to salbutamol, six of these β-agonists showed more inhibition (83 to 66%) in the ELISA after IF and the other four β-agonists showed less inhibition (60 to 40%), which could be explained by the cross-reactivity of the anti-salbutamol antibodies3 and the β-agonist-ELISA6 for these compounds.| β-Agonist | Inhibition (%) | |||
|---|---|---|---|---|
| ELISA | IF-ELISA | |||
| 1 ng ml−1 | 5 ng ml−1 | 1 ng ml−1 | 5 ng ml−1 | |
| a Nt = not tested. | ||||
| Cimbuterol | 86 | 96 | 83 | 91 |
| Clenbuterol | 82 | 96 | 78 | 94 |
| Bromobuterol | 81 | 95 | 80 | 91 |
| Mabuterol | 79 | 93 | 77 | 98 |
| Mapenterol | 75 | 93 | 67 | 88 |
| Salbutamol | 69 | 84 | 64 | 84 |
| Tulobuterol | 67 | 83 | 66 | 81 |
| Terbutaline | 58 | 79 | 40 | 68 |
| Clenproperol | 56 | 76 | 55 | 74 |
| Carbuterol | 55 | 83 | 58 | 74 |
| Cimaterol | 56 | 76 | 55 | 74 |
| Pirbuterol | 48 | 75 | 18 | 50 |
| Metaproterenol | 15 | 39 | 6 | 8 |
| Fenoterol | <10 | <10 | Nta | Nt |
| Ractopamine | <10 | <10 | Nt | Nt |
| Clenpenterol | <10 | <10 | Nt | Nt |
| Isoxsuprine | <10 | <10 | Nt | Nt |
| Clencyclohexerol | <10 | <10 | Nt | Nt |
Comparing the results obtained in the ELISA (with and without IF) for the eleven β-agonists at the 1 ng ml−1 level (expressed as salbutamol equivalents), the mean concentration found with IF-ELISA was only 10% lower than found with the ELISA.
Pirbuterol and metaproterenol showed only low inhibition in the ELISA after IF and, because of the low cross-reactivity for five other β-agonists, these were not tested in IF-ELISA (see Table 1).
Incurred urine samples
Incurred urine samples selected from The Netherlands National Programme (taken in the years 2000 and 2001), in which low levels of salbutamol were previously detected and confirmed by GC-MS, were re-analysed with IF in combination with the β-agonist ELISA (see Table 2). All samples showed concentrations above the LOD of the screening procedure (>0.14 ng ml−1) and also above the limit of determination for the method (0.22 ng ml−1). The average concentration found for the sample screening (1.1 ng ml−1) was 30% lower than the average concentration found by GC-MS (1.6 ng ml−1). This could be largely explained by the average recovery after IF (77%), while in the GC-MS procedure an internal standard (salbutamol-d6) was used to correct for the recovery.| Sample No. | Year of sampling | GC-MS salbutamol/ng ml−1 | IF-ELISA salbutamol equivalents/ng ml−1 |
|---|---|---|---|
| 1064 | 2001 | 1.3 | 1.0 ± 0.3 |
| 1065 | 2001 | 2.3 | 1.5 ± 0.6 |
| 1066 | 2001 | 2.6 | 1.5 ± 0.4 |
| 6604 | 2000 | 1.1 | 1.3 ± 0.0 |
| 6605 | 2000 | 0.6 | 0.4 ± 0.0 |
| 6606 | 2000 | 1.8 | 1.4 ± 0.4 |
| 6607 | 2000 | 1.4 | 1.1 ± 0.3 |
| 6608 | 2000 | 2.2 | 1.0 ± 0.1 |
| 6705 | 2000 | 2.5 | 0.8 ± 0.0 |
| 6726 | 2000 | 1.1 | 1.2 ± 0.1 |
| 6727 | 2000 | 0.8 | 1.1 ± 0.3 |
| 6728 | 2000 | 0.9 | 0.8 ± 0.1 |
| 6729 | 2000 | 2.3 | 1.1 ± 0.1 |
Salbutamol is excreted in urine mainly as the glucuronide or sulfate derivative.10 The positive screening results obtained with these incurred urine samples (without deconjugation) prove that the anti-salbutamol antibodies react with these derivatives and, as a result of this, a deconjugation step can be omitted during sample cleanup.
Further incurred urine samples containing low levels of four other β-agonists were analysed by IF-ELISA and the results were compared with GC-MS (see Table 3). All samples showed concentrations above the LDM of the screening procedure (>0.22 ng ml−1), which proves that the procedure works for the detection of these β-agonists in incurred urine samples.
| Sample No. | Year of sampling | β-Agonist | GC-MSa/ng ml−1 | GC-MSb/ng ml−1 | IF-ELISAb salbutamol equivalents/ng ml−1 |
|---|---|---|---|---|---|
| a Analysed directly after receipt of the sample. b Re-analysed in May 2001. | |||||
| 698 | 1997 | Clenbuterol | 0.3 | 0.3 | 0.9 |
| 4680 | 1996 | Clenbuterol | 1.0 | 1.1 | 2.0 |
| 5138 | 1996 | Clenbuterol | 0.6 | 0.7 | 2.0 |
| 5744 | 1995 | Clenbuterol | 1.2 | 1.6 | >5 |
| 7071 | 1995 | Clenbuterol | 1.2 | 1.6 | >5 |
| 228 | 1994 | Clenbuterol | 4.7 | >5 | >5 |
| 3807 | 1994 | Clenbuterol | 1.1 | 1.1 | >5 |
| 46 | 1997 | Bromobuterol | 0.6 | 0.8 | 0.7 |
| 746 | 1993 | Bromobuterol | 3.1 | >5 | >5 |
| 1001 | 1993 | Mabuterol | 0.5 | 0.9 | 0.8 |
| 707 | 1991 | Mabuterol | 3.1 | 4.5 | >5 |
| 222 | 1992 | Mapenterol | 0.6 | 0.7 | 0.5 |
| 415 | 1992 | Mapenterol | 0.3 | 0.6 | 0.8 |
| 670 | 1992 | Mapenterol | 1.0 | 2.5 | >5 |
| 1316 | 1991 | Mapenterol | 1.0 | 2.0 | 1.5 |
| 1317 | 1991 | Mapenterol | 0.5 | 1.1 | 1.7 |
| 1391 | 1991 | Mapenterol | 1.8 | 1.8 | 1.7 |
| 1392 | 1991 | Mapenterol | 1.9 | 1.9 | >5 |
| 2482 | 1991 | Mapenterol | 1.5 | 1.8 | 1.4 |
| 2483 | 1991 | Mapenterol | 1.3 | 1.4 | 0.9 |
| 2485 | 1991 | Mapenterol | 1.1 | 1.3 | 2.1 |
The concentrations of clenbuterol found by IF-ELISA were higher than those obtained with GC-MS. This could be explained partly by the higher cross-reactivity of clenbuterol, compared with salbutamol, in the β-agonist ELISA. Another explanation might be the possible presence of polar metabolites of clenbuterol in the urine14 which might react with the antibodies and which were not detected by GC-MS. Such differences were not observed for bromobuterol, mabuterol and mapenterol in the incurred urine samples.
The incurred urine samples were stored for four to ten years at −20 °C after the first analyses by GC-MS. Re-analysing those urine samples after four to ten years still resulted in comparable concentrations (see Table 3) which proved the stability of these β-agonists in urine stored under these conditions.
Comparison of IF-ELISA with isobutanol extraction combined with ELISA
During the last few years, combination of an isobutanol extraction (see Experimental) with the β-agonist ELISA was used for the screening of urine samples from The Netherlands National Programme with an action level of 0.4 ng ml−1 (salbutamol equivalents). Although this worked successfully for some years, recently many false positive results were obtained with bovine urine samples taken at the farm. The cause of this is still unknown and this observation was one of the reasons to look for a new sample cleanup. Some of these samples were re-analysed by IF-ELISA and the results are compared in Table 4. The average concentration found by IF-ELISA (0.12 ng ml−1) was approximately six times lower than that found by the extraction procedure in combination with the ELISA (0.68 ng ml−1). In seven of these samples, levels above the LOD (>0.14 ng ml−1) of the IF-ELISA procedure were found (0.15–0.21 ng ml−1). This might be explained by the possible presence of low levels of β-agonists which were below the LODs of the GC-MS procedure (0.2 to 1 ng ml−1, depending on the β-agonist concerned). However, the concentrations found by IF-ELISA in these samples were all below the LDM of 0.22 ng ml−1, which means that the new screening procedure resulted in less false positive results compared with the former screening procedure.| Sample No. | Year of sampling | ELISA + extractiona salbutamol equivalents/ng ml−1 | IF-ELISA salbutamol equivalents/ng ml−1 |
|---|---|---|---|
| a Extraction with isobutanol (see Experimental). | |||
| 0089 | 2001 | 0.90 | 0.08 |
| 0091 | 2001 | 0.93 | 0.21 |
| 0092 | 2001 | 0.70 | 0.16 |
| 0722 | 2001 | 0.58 | 0.12 |
| 0723 | 2001 | 0.74 | 0.15 |
| 0724 | 2001 | 0.84 | 0.12 |
| 0725 | 2001 | 0.87 | 0.19 |
| 0726 | 2001 | 0.86 | 0.12 |
| 1063 | 2001 | 0.36 | 0.05 |
| 5086 | 2000 | 0.51 | 0.10 |
| 6243 | 2000 | 0.75 | 0.11 |
| 6246 | 2000 | 0.82 | 0.15 |
| 6247 | 2000 | 0.64 | 0.18 |
| 7163 | 2000 | 0.64 | 0.09 |
| 7165 | 2000 | 0.73 | 0.04 |
| 7166 | 2000 | 0.85 | 0.16 |
| 7433 | 2000 | 0.48 | 0.10 |
| 7437 | 2000 | 0.46 | 0.08 |
| 7524 | 2000 | 0.61 | 0.08 |
| 7525 | 2000 | 0.65 | 0.10 |
| 7526 | 2000 | 0.50 | 0.11 |
| 7527 | 2000 | 0.60 | 0.11 |
References
- Official Journal of the European Communities (1996), Council Directive 96/22/EC, No. L 125/3-9.
- H. A. Kuiper, M. Y. Noordam, M. M. H. van Dooren-Flipsen, R. Schilt and A. H. Roos, J. Anim. Sci., 1998, 76, 195 Search PubMed.
- W. Haasnoot, G. Cazemier, P. Stouten and A. Kemmers-Voncken, in Immunoassays for Residue Analyis, Food Safety, ed. R. C. Beier and L. H. Stanker, American Chemical Society, Washington DC, 1996, pp. 60–73 Search PubMed.
- R. Schilt, W. Haasnoot, M. A. Jonker, H. Hooijerink and R. J. A. Paulussen, in Euro-Residue Conference, Proceedings of an International Conference, Noordwijkerhout, The Netherlands, ed. N. Ruiter and P. B. Czedik-Eysenberg, Rijksuniversiteit Utrecht, Faculteit der Diergeneeskunde, Utrecht, The Netherlands, 1990, p. 320 Search PubMed.
- J. A. van Rhijn, W. A. Traag and H. H. Heskamp, J. Chromatogr., 1993, 619, 243 CrossRef CAS.
- W. Haasnoot, L. Streppel, G. Cazemier, M. Salden, P. Stouten, M. Essers and P. van Wichen, Analyst, 1996, 121, 1111 RSC.
- S. A. Haughey, G. A. Baxter, C. T. Elliott, B. Persson, C. Jonson and P. Bjurling, J. AOAC Int., 2001, 84, 1025 Search PubMed.
- W. Haasnoot, P. Stouten, R. Schilt and H. Hooijerink, Analyst, 1998, 123, 2707 RSC.
- K. E. I. Vanoosthuyze, C. J. M. Arts and C. H. Van Peteghem, J. Agric. Food Chem., 1997, 45, 3129 CrossRef CAS.
- R. F. Witcamp, in Proceedings of Scientific Conference on Growth Promoters in Meat Production, Brussels, 29th November–1st December 1995, Office for Official Publications of the European Communities, Luxembourg, 1996, pp. 297–315 Search PubMed.
- K.-E. Hellenäs, M. A. Johansson, C. T. Elliott, A. Sternesjö and E. Stenberg, in EuroResidue IV. Proceedings of the Conference on Residues of Veterinary Drugs in Food, ed. L. A. van Ginkel and A. Ruiter, RIVM, Laboratory for residue analysis, Bilthoven, The Netherlands, 2000, pp. 542–545 Search PubMed.
- W. Haasnoot, A. Kemmers-Voncken, H. van Rhijn and R. Schilt, in EuroResidue IV. Proceedings of the Conference on Residues of Veterinary Drugs in Food, ed. L. A. van Ginkel and A. Ruiter, RIVM, Laboratory for Residue Analysis, Bilthoven, The Netherlands, 2000, pp. 496–500 Search PubMed.
- S. S. Sterk, E. F. van Tricht, R. W. Stephany and L. A. van Ginkel, RIVM report 3890002 047, RIVM, Bilthoven, The Netherlands, 1998 Search PubMed.
- D. Zalko, G. Bories and J. Tulliez, J. Agric. Food Chem., 1988, 46, 1935 CrossRef.
| This journal is © The Royal Society of Chemistry 2002 |