Determination of furosemide in urine samples by direct injection in a micellar liquid chromatographic system
S.
Carda-Broch
a,
J.
Esteve-Romero
b,
M. J.
Ruiz-Angel
a and
M. C.
García-Alvarez-Coque
*a
aDepartament de Química Analítica, Facultat de Química, Universitat de València, Dr. Moliner 50, 46100, Burjassot (València), Spain
bArea de Química Analítica, Universitat Jaume I, Cra. Borriol s/n, 12080, Castelló, Spain
First published on 5th December 2001
Abstract
A sensitive, selective and efficient micellar liquid chromatographic (MLC) procedure was developed for the determination of furosemide (4-chloro-N-furfuryl-5-sulfamoylanthranilic acid) in urine samples by direct injection and UV detection. The procedure makes use of a C18 reversed-phase column and a micellar mobile phase of 0.05 mol l−1 sodium dodecyl sulfate–6% v/v propanol and phosphate buffer at pH 3 to resolve furosemide from its photochemical degradation products. The importance of protecting the standards and urine samples to be analysed from light in the assay of furosemide, avoiding its degradation, was verified. The limit of quantification was 0.15 μg ml−1 and the relative standard deviation of the inter-day assay was 0.8–0.04% in the 6–82 μg ml−1 range. Detection of urinary excretion of furosemide was followed up to 12 h after ingestion of the drug by a healthy volunteer. No potential interference from the major metabolite (furosemide acylglucuronide) and its hydrolytic product (4-chloro-5-sulfamoylanthranilic acid) was observed. Commonly administered drugs also did not interfere. The proposed MLC procedure permits the rapid and reproducible measurement of low levels of furosemide in a small amount of urine.
Introduction
Furosemide, an anthranilic acid derivative (see Fig.1), is a potent diuretic that inhibits the active reabsorption of chloride in the diluting segment of the loop of Henle, thus preventing the reabsorption of sodium, which passively follows chloride.1 This loop diuretic is commonly used for the treatment of renal diseases, congestive heart failure and hypertension.2 It is normally administered as tablets and intravenous or intramuscular injections. The drug is capable of initiating adverse light-induced biological effects and has been found to produce photosensitivity, phototoxicity, oxygen-dependent photohemolysis and lipid photoperoxidation.3 Furosemide is mainly excreted through the kidneys, but the faecal recovery of 7–9% of intravenously administered drug shows that the diuretic or its metabolites are also excreted by other routes. | ||
| Fig. 1 Initial degradation of furosemide. | ||
Furosemide has been banned in sport by the Medical Commission of the International Olympic Commitee since 1986, because of the increase in urine flow, which reduces body weight, especially in sports where competition is based on weight classes.4 Owing to the diluting effect, the intake of this kind of drug could also result in suppression of other doping substances below their limits of detection.5
Photochemical degradation of aqueous and methanolic solutions of furosemide under the influence of UV radiation has been reported by several workers. In acidic solution, rapid degradation takes place.3,6–10 Furosemide seems to undergo photooxidation, photohydrolysis and photodechlorination.6,10 At high temperature, furosemide is hydrolysed to 4-chloro-5-sulfamoylanthranilic acid (CSA) and furfuryl alcohol,9,11,12 which is quickly converted into levulinic acid (Fig. 1).13,14 The degradation may be even faster if sulfate ion is present,15 as in normal urine. Urine is usually weakly acidic and may be made more acidic by the action of furosemide.16 In contrast, in alkaline solution, furosemide has been reported to show high stability.14,15,17 However, in humans, furosemide is metabolised to its acylglucuronide,18 which is unstable in alkaline media; therefore, urine has been recommended to be kept acidic at pH 5 for analysis in order to prevent the hydrolysis and isomerisation of this metabolite.19–23 Beermann et al.18 found no evidence of furosemide degradation in the upper digestive tract after analysing gastrointestinal aspirates and that furosemide was stable when incubated in gastric or duodenal juice, bile or urine for up to 2 h.
Rapid and reliable methods are required for the determination of furosemide in therapeutic drug monitoring and doping control, especially for urine samples because the action of the diuretic seems to correlate best with its urinary levels.24–26Different methods using high-performance liquid chromatography (HPLC) to measure furosemide have been described.27–33 Degradation problems with urine samples are rarely mentioned in analytical reports. Potential interference from the major metabolite, furosemide acylglucuronide or the degradation products has not been considered for the majority of analytical methods. This may limit their usefulness in the therapeutic monitoring of furosemide at the levels found in patients, and also in pharmacokinetic bioequivalence studies of the drug.
In a previous study,34 we demonstrated that furosemide solutions are stable in a micellar medium of sodium dodecyl sulfate (SDS) at pH 3–5 protected from light. These conditions should be used to keep furosemide standard solutions and samples in the analytical laboratory. We reported a chromatographic procedure with micellar mobile phases containing SDS, which was applied to the assay of the diuretic in numerous pharmaceuticals commercialised in several dosage forms. Under the optimised experimental conditions furosemide was resolved from its photodegradation products.
Since furosemide suffers photodegradation, it is necessary to protect urine samples from light to avoid partial or total decomposition of the drug. When a sportsman deliberately takes furosemide, it is possible that the drug will remain undetected if adequate precautions are not taken. The aim of this work was the application of a rapid, sensitive and selective reversed-phase HPLC procedure with micellar mobile phases to determine furosemide in urine samples, which avoids the degradation of the drug in the matrix.
Experimental
Reagents
Furosemide and furfuryl alcohol standards were obtained from Sigma (St. Louis, MO, USA). 4-Chloro-5-sulfamoylanthranilic acid was kindly supplied by Hoechst (Frankfurt-am-Main, Germany). Since the drug is only sparingly soluble in water, it was first dissolved in a small amount of ethanol (the final ethanol concentration was 10%) and further diluted with SDS (99% purity, Merck, Darmstadt, Germany) solution. Other reagents were sodium dihydrogenphosphate, disodium hydrogenphosphate (for analysis, Panreac, Barcelona, Spain), HCl, NaOH (Probus, Badalona, Spain), methanol, propan-1-ol (HPLC grade, Scharlab, Barcelona, Spain) and ethanol (for analysis, Prolabo, Paris, France). Nanopure water (Barnstead, Sybron, Boston, MA, USA) was used to prepare aqueous solutions.Seguril (Hoechst Pharma, S. Feliu de Llobregat, Barcelona, Spain), Furosemide EG (Eurogenerics, Brussels, Belgium) and Furosémide RPG (Laboratoires Biogaléniques, Paris, France) were administered to a healthy volunteer on different days to obtain urinary excretion curves for furosemide.
Apparatus and chromatographic conditions
The HPLC system consisted of a Series HP 1100 chromatograph (Agilent, Palo Alto, CA, USA), provided with an isocratic pump (Model G1310A), an autosampler with 2 ml vials (Model G1313A) and a UV–visible diode-array detector (Model G1315A). A PC workstation with HP3D software was used for instrumental control and acquisition of chromatographic data. MICHROM software was used for optimisation studies.35The analytical separation was accomplished using an ODS-2 reversed-phase column (5 μm particle size, 125 × 4.6 mm id) (Scharlab), that was connected to a 30 mm guard precolumn of similar characteristics (Scharlab). The flow rate was 1.0 ml min−1 and the injection volume was 20 μl. The chromatographic runs were carried out at laboratory temperature. Monitoring was performed at 274 nm.
The micellar mobile phase used to resolve the mixture of furosemide and its photodegradation products was 0.05 mol l−1 SDS–6% propanol–0.01 mol l−1 NaH2PO4 at pH 3. The pH was buffered before the addition of propanol to the micellar solution. The mobile phases were filtered through 0.45 μm nylon membranes of 47 mm diameter (Micron Separations, Westboro, MA, USA).
Standard solutions and urine samples
Stock standard solutions containing 100 μg ml−1 of furosemide were prepared. Furosemide was dissolved in 10 ml of ethanol with the aid of an ultrasonic bath (Model 617, Selecta, Barcelona, Spain) and was made up to the mark in a 100 ml calibrated flask with 0.10 mol l−1 SDS buffered at pH 3 with phosphate. For the stability studies urine was spiked with furosemide solution and diluted with 0.10 mol l−1 SDS to a final concentration of 10 μg ml−1.The analyses were performed with 1 ml urine samples, which were diluted 1∶25 with 0.10 mol l−1 SDS at pH 3 before injection. All solutions were protected from light by covering them with aluminium foil and kept in the dark at 4 °C, to avoid photochemical degradation of the drug. The urine samples containing spiked or excreted furosemide were filtered before their injection into the chromatograph. However, the filtration was always performed directly into the autosampler vials through 0.45 μm nylon membranes of 13 mm diameter (Micron Separations). The transparent glass vials were also protected from light. The optimisation of the procedure was performed with spiked urine samples.
Results and discussion
Optimisation of the separation of furosemide and its photodegradation products
In previous work, the best chromatographic conditions to resolve furosemide from its photodegradation products were studied.34 The analysis of furosemide samples was carried out with mobile phases of micellar SDS buffered at pH 3–5, since the retention times and plate counts were excessively low at higher pH. The chromatograms of furosemide aqueous solutions exposed to artificial light contain several peaks at retention times shorter than that of the peak of furosemide, which were assigned to different degradation products, based on the relative retentions, absorption wavelengths and the information found in the literature about their nature: CSA, furfuryl alcohol (I), a CSA degradation product where the carboxylic group is released (II) and a furosemide dechlorination product (presumably N-furfuryl-5-sulfamoylanthranilic acid) (III).34 The degradation products could not be identified by direct coupling of micellar liquid chromatography (MLC) with mass spectrometry, owing to the high concentration of surfactant in the mobile phase. The same peaks were observed for urine samples containing spiked or excreted furosemide exposed to artificial light, with the exception of CSA, the major degradation product of furosemide, the retention time of which was less than 2 min and was completely overlapped by the broad band originated by the proteins in urine. Upon direct sunlight irradiation, another peak appears in furosemide aqueous solutions close to the peak of furosemide, formed from the diuretic lost of the carboxylic group. This peak was not observed in spiked urine samples after 6 d of exposure to sunlight.To optimise the separation of furosemide from its degradation products in urine, a mixture of degraded drug was eluted with several mobile phases containing SDS at pH 3 and a small amount of propanol (not greater than 8% v/v). The concentration of SDS was in the 0.04–0.14 mol l−1 range. Fig. 2 shows the global resolution surface obtained for this separation. A wide region of optimal resolution is observed in the 0.04–0.06 mol l−1 SDS concentration range and the whole range of propanol concentrations. A much narrower (less robust) region of maximum resolution was obtained close to 0.10 mol l−1 SDS–1% propanol.
 | ||
| Fig. 2 Global resolution diagram for the separation of furosemide and its degradation products. | ||
A mobile phase containing 0.05 mol l−1 SDS and 6% propanol was selected for the stability studies and to carry out the determination of furosemide in urine samples. This mobile phase elutes the drug in a short time (6.4 min) and permits monitoring its most significant photodegradation products without any interference from the endogenous compounds. Fig. 3 shows a chromatogram of furosemide in a urine solution degraded after exposure to sunlight at different times
 | ||
| Fig. 3 Chromatogram of furosemide and its photodegradation products in a spiked urine solution protected from (a) or exposed to sunlight for 20 min (b) and 8 h (c). | ||
Stability of furosemide solutions
The acid-catalysed degradation of furosemide is slower in micellar solution than aqueous medium.34 The acidic solutions of furosemide protected from light are highly stable, but when exposed to light decompose at greater rate at pH 3 than at pH 5. However, at pH >5 the chromatographic peak of furosemide is shifted progressively to shorter retention times and its quantification is more problematic. In urine, this effect is observed at pH >4. For this reason, the pH of the assayed urine samples should be maintained between 3 and 4.The stability of furosemide in urine samples was next studied at pH 3. Several workers have suggested that low pH causes degradative breakdown of furosemide in urine.28,29 However, according to Singh et al.,29 furosemide was observed to be stable at acidic pH under the experimental conditions used in this work. On the other hand, the direct injection of a large number of urine samples can shorten the life of the chromatographic column or can force frequent regeneration of the stationary phase. Therefore, the dilution of urine samples is convenient, although the injection of undiluted samples to reach lower limits of detection is feasible.
Spiked urine samples were prepared by mixing one volume of urine and 25 volumes of a solution of the drug buffered at pH 3 and kept from light or exposed to standard laboratory lighting (Osram 40 W fluorescent light at 1.5 m), at room temperature. No precautions were taken to prevent contact of the samples with air. Decomposition of furosemide obeys first-order kinetics.9,10,27 The logarithm of the difference between the area at any time, At and the final area of the chromatographic peak, A∞ (which should be zero), are plotted against time in Fig. 4, for urine spiked samples kept from light or exposed to artificial light. The apparent first-order rate constants were obtained from the slopes of the linear segment according to the equation ln(At − A∞) = ln(A0 − A∞) − k1t where A0 is the area at time zero and k1 the apparent first-order rate constant. The solution protected from light (Fig. 4, line a) was stable during long periods, whereas that unprotected (Fig. 4, line b) suffered photodegradation with a half-life of 22 d. The peaks of the degradation products were not observed in the spiked urine solutions protected from light, at least after 9 d from the preparation. After 3 d, the area of the peak of furosemide protected from light decreased by only 0.4%, whereas for the unprotected solutions it decreased by 10%. When these solutions were directly exposed to sunlight, the diuretic was completely decomposed after 3 h, with a half-life of 30 min.
 | ||
| Fig. 4 First-order plots for the degradation of furosemide in a urine matrix at pH 3, protected from light (a) and exposed to artificial laboratory light at room temperature (b). The concentration of furosemide was 10 μg ml−1. | ||
Analysis of urine samples
The analytical figures of merit were obtained using drug-free urine samples collected from healthy adult volunteers (men and women). The matrix and spiked samples were analysed using a mobile phase of 0.05 mol l−1 SDS–6% propanol at pH 3. Several published furosemide assays failed to report the possible interferences from furosemide metabolites and degradation products. This lack of specificity may result in a wrong estimation of actual concentrations, leading to erroneous bioavailability/bioequivalence decisions. The composition and pH of the mobile phase used in the assay proposed in this work provided a good separation of furosemide from its degradation products and no interference from endogenous components in urine or furosemide glucuronide (no peak was found that could be assigned to this metabolite). This allows the control of the photodegradation of furosemide in the standards and samples during the analytical process.To evaluate the accuracy and precision of the procedure, a urine matrix was spiked with five different known concentrations of the drug. The spiked samples were analysed on the same day and on different days up to 6 d to determine the variability (Table 1). Each level of concentration was injected sixfold every day. The within-day precision showed a relative standard deviation (RSD) of up to 0.19% and the day-to-day precision was <0.8%.
| Added concentration/ μg ml−1 | Within-daya | Day-to-daya | ||||
|---|---|---|---|---|---|---|
| Measured concentration/ μg ml−1 | Accuracy (%) | Precision (%) | Measured concentration/ μg ml−1 | Accuracy (%) | Precision (%) | |
| a n = 6. | ||||||
| 6.1 | 6.196 ± 0.005 | 1.2 | 0.08 | 6.14 ± 0.05 | 0.3 | 0.8 |
| 10.2 | 10.26 ± 0.02 | 0.6 | 0.19 | 10.20 ± 0.05 | 0 | 0.5 |
| 20.4 | 20.300 ± 0.017 | −0.5 | 0.08 | 20.27 ± 0.03 | −0.6 | 0.15 |
| 51.0 | 51.091 ± 0.004 | 0.2 | 0.008 | 51.10 ± 0.05 | 0.10 | 0.2 |
| 81.6 | 81.578 ± 0.005 | −0.03 | 0.006 | 81.55 ± 0.03 | −0.06 | 0.04 |
The same furosemide spiked standards at five increasing concentrations in the range 1–21 μg ml−1 were injected during several days to obtain the variability of the parameters of the calibration curve (Table 2). The solutions were kept in the dark at 4 °C between series. Calibration curves were constructed using the measured areas of the chromatographic peaks of duplicate injections. The results in Table 2 show an acceptable linearity (r > 0.9999) over the studied concentration range. The intercepts are statistically zero and the RSD of the slopes is 0.4%. Therefore, the calibration standards can be used during at least 10 d without observing significant degradation when kept from light.
| Day | Intercept | Slope | r |
|---|---|---|---|
| a The first six calibration curves were obtained during six consecutive days. | |||
| 1 | 0.1 ± 0.3 | 1.06 ± 0.02 | 0.99998 |
| 2 | 0.0 ± 0.2 | 1.051 ± 0.018 | 0.99996 |
| 3 | 0.2 ± 0.3 | 1.05 ± 0.03 | 0.99991 |
| 4 | 0.2 ± 0.3 | 1.05 ± 0.02 | 0.99994 |
| 5 | 0.2 ± 0.3 | 1.05 ± 0.02 | 0.99994 |
| 6 | 0.12 ± 0.15 | 1.055 ± 0.012 | 0.99998 |
| 10 | 0.1 ± 0.3 | 1.05 ± 0.03 | 0.99990 |
| Mean | 1.052 ± 0.004 | ||
Five calibration curves were further constructed using furosemide standards prepared on different days during several months, at 6–9 concentrations in the range 0.4–100 μg ml−1, to determine the variability of the intercepts and slopes between independent calibration curves. The results in Table 3 show the small day-to-day variability of the slopes, with an RSD of 1.8%. A calibration curve was also constructed with furosemide standards prepared in aqueous micellar solution and compared with a curve obtained on the same day with spiked urine standards. The intercepts were 0.19 and −0.16 and the slopes 1.113 and 1.104 for the furosemide aqueous and urine standards, respectively, with r = 0.999 (n = 9). The difference between the slopes is similar to that obtained between days for urine spiked samples, which indicates the absence of matrix effects. The limit of quantification for furosemide, defined as the analyte concentration producing a peak area equal to 10s (s = standard deviation of the peak area of the analyte at a low concentration) was 0.08 and 0.15 μg ml−1 for aqueous and urine solutions, respectively.
| Day | Intercept | Slope | r |
|---|---|---|---|
| a Assays performed during several months. | |||
| 1 | −0.16 ± 0.19 | 1.104 ± 0.004 | 0.99999 |
| 2 | 0.1 ± 0.3 | 1.06 ± 0.02 | 0.99998 |
| 3 | 0.14 ± 0.10 | 1.119 ± 0.003 | 0.999997 |
| 4 | 0.28 ± 0.08 | 1.076 ± 0.007 | 0.99999 |
| 5 | 0.01 ± 0.10 | 1.090 ± 0.008 | 0.999991 |
| Mean | 1.09 ± 0.02 | ||
Finally, a calibration curve constructed with protected furosemide standards in urine matrix was compared with another prepared on the same day using a furosemide stock standard solution and diluted standards exposed to light. In both cases, the time from the preparation of the solutions to the last injection was ∼100 min. The intercepts were 0.28 and −0.61 and the slopes 1.076 and 0.925 for the protected and unprotected standards, respectively, with r = 0.999 (n = 6). This means a 14% decrease in the slope of the calibration curve for the unprotected solutions, which can yield a considerable error in the quantification of the drug.
To determine the reliability of the assay, 22 urine samples from different volunteers were used. The matrix samples (in the absence of furosemide) were chromatographed using the proposed procedure. Chromatograms were inspected for the presence of interfering peaks from endogeneous compounds. No interference was found with the peak of furosemide at 6.4 min. Moreover, several commonly administered drugs were tested for their possible interference under the selected chromatographic conditions. Table 4 lists the retention times of these drugs with the 0.05 mol l−1 SDS–6% propanol mobile phase. None of the assayed compounds yielded chromatographic peaks that interfered in the analyses.
| Drug | Retention time/mina |
|---|---|
| a N.D., not detected. | |
| Acetylsalicylic acid | 3.4 |
| N-Acetyl-L-cysteine | N.D. (after 21 min) |
| Amiloride | 16.2 |
| Amoxicillin | 8.1 |
| Ascorbic acid | 1.2 |
| Atenolol | 13.4 |
| Bendroflumethiazide | 7.2 |
| Caffeine | 5.0 |
| Captopril | N.D. (274 nm); 2.7 (225 nm) |
| Carbamazepine | 14.2 |
| Chlorpheniramine | 1.1 |
| Cocaine | 87.7 |
| Codeine | 27.8 |
| Diazepam | 38.8 |
| Diltiazem | 69.9 |
| Hydralazine | 31.0 |
| Hydrochlorothiazide | 1.7 |
| Metoclopramide | 91.2 |
| Metoprolol | 44.4 |
| Paracetamol | 1.7 |
| Propranolol | 72.2 |
| Salbutamol | 12.9 |
| Spironolactone | 46.7 |
| Sulfamethoxazole | 4.6 |
| Triamterene | 25.9 |
| Xanthinol | 16.6 |
The procedure was applied in a pharmacokinetic study to demonstrate its usefulness. A urinary excretion study was performed with a healthy volunteer who ingested a 40 mg furosemide tablet from three manufacturers on different days. Each time, a sample was collected just before the administration of the drug to be used as the blank. Other urine samples were collected at appropriate time intervals post-dose, protected from light and refrigerated at 4 °C until analysed. To avoid dehydration and electrolyte depletion, the subject drank water during the study. Vree et al.36 made the urinary pH acidic (pH 5.0–5.5) by the oral intake of 1 g of ammonium chloride four times per day to perform a similar analysis. In our case, the urinary pH was not changed.
Fig. 5(a) shows the urine concentration profile of furosemide obtained on different days for the same individual and Fig. 5(b) shows the cumulative amounts of unchanged furosemide in urine. The dose excreted unchanged for furosemide each day was 14.5, 15.9 and 16.0%. Peaks which could be assigned to metabolites of the diuretic or degradation products were not observed in any case. Furosemide could be detected up to 12 h from its ingestion.
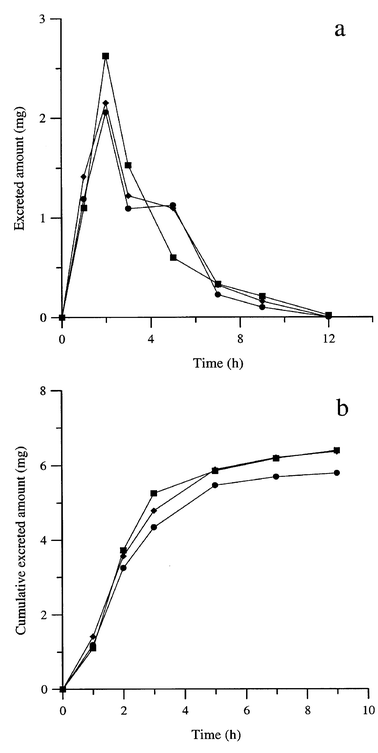 | ||
| Fig. 5 Urine concentration profiles of furosemide after oral administration of a 40 mg tablet of three formulations (a) and cumulative amounts of unchanged furosemide in urine (b). | ||
When a urine sample obtained 3 h after the drug intake was exposed to direct sunlight, the concentration of furosemide decreased by 44% after 10 min and by 79% after 30 min. After 1 hour, the signal was almost zero. These results show the importance of protecting the analysed urine samples from light in the assay of furosemide. If no care is taken and the analyses are performed several hours after the collection, no evidence will remain of the use or abuse of this drug.
Conclusions
The studies performed in this work show that furosemide is stable in urine samples at pH 3 when protected from light. When exposed to light, the degradation is complex, giving rise to several products. The proposed chromatographic procedure gives good results for the determination of furosemide in urine, in terms of its accuracy, repeatability and selectivity. Other commonly administered drugs do not interfere and the limit of detection is at the ng ml−1 level. No potential interference from the major furosemide metabolite, furosemide acylglucuronide, and its hydrolytic product, CSA, was observed. This makes the proposed procedure particularly useful for application in pharmacokinetic studies of healthy subjects and patients, using small volumes of urine samples.The proposed procedure is also simpler than most methods reported for this drug that use aqueous–organic mobile phases, where a previous extraction and an internal standard are recommended.28,29,31,32 After dilution of the urine sample this treatment can be avoided, as proposed by Farthing et al.30 and Nava-Ocampo et al.33 However, these authors recommended the use of a guard column (where the samples were injected), which should be replaced every 100 injections to prevent column degradation, or centrifugation of the sample and filtration through filter-paper before injection. Although dilution of the sample is recommended in the proposed micellar chromatographic procedure, direct injection of the sample into the analytical column without any dilution is possible. In the literature, a mobile phase of 30–35% v/v acetonitrile is often used to achieve a retention time of 9–10 min for furosemide eluted from C18 columns. Nava-Ocampo et al.33 replaced methanol with acetonitrile to decrease the cost and toxicity of the mobile phase. The amount of organic solvent is significantly lower in the proposed micellar mobile phase: 6% propanol elutes furosemide in 6.4 min, but a lower concentration of this solvent (2%) can be also used, producing a retention time of ∼11 min. Propanol is also less toxic than methanol and is highly retained in the SDS micellar solution, reducing the risk of evaporation.
Acknowledgements
This work was supported by the DGICYT, Project PB97/1384 (Spain), the MCYT Project BQU2001–3047 and the Bancaixa, Project P1A97/16 (Spain). M. J. Ruiz-Angel thanks the Ministerio de Ciencia y Tecnología of Spain for an FPI grant.References
- H. R. Jacobson and J. P. Kokko, Annu. Rev. Pharmacol. Toxicol., 1976, 16, 201 CrossRef CAS.
- D. C. Brater, Drugs, 1991, 41, 14 Search PubMed.
- F. Vargas, I. Martínez-Volkmar, J. Sequera, H. Méndez, J. Rojas, G. Fraile, M. Velásquez and R. Medina, J. Photochem. Photobiol., B, 1998, 42, 219 Search PubMed.
- C. Rodríguez-Bueno, A. F. Rodríguez-Carreras, J. I. Maynar, R. Cortés and D. Maynar, M. Arch. Med. Deporte, 1991, 3, 411 Search PubMed.
- C. March, D. Farthing, B. Wells, E. Besenfelder and H. T. Karnes, J. Pharm. Sci., 1990, 79, 453 CAS.
- P. C. Rowbotham, J. B. Stanford and J. K. Sugden, Pharm. Acta Helv., 1976, 51, 304 CAS.
- D. E. Moore and V. Sithipitaks, J. Pharm. Pharmacol., 1983, 35, 489 Search PubMed.
- A. M. Yahya, J. C. McElnay and P. F. D’Arcy, Int. J. Pharm., 1986, 31, 65 CrossRef CAS.
- H. Bundgaard, T. Norgaard and N. M. Nielsen, Int. J. Pharm., 1988, 42, 217 CrossRef CAS.
- A F. Asker and A. J. Ferdous, PDA J. Pharm. Sci. Technol., 1996, 50, 158 Search PubMed.
- K. Sturm, W. Siedel, R. Weyer and H. Ruschig, Chem. Ber., 1966, 99, 328 Search PubMed.
- J. E. Cruz, D. D. Maness and G. J. Yakatan, Int. J. Pharm., 1979, 2, 275 CrossRef CAS.
- K. A. Kovar, G. P. Wojtovicz and H. Auterhoff, Arch. Pharm. (Weinheim, Ger.), 1974, 307, 657 Search PubMed.
- A. G. Ghanekar, V. D. Gupta and C. W. Gibbs, J. Pharm. Sci., 1978, 67, 808 CAS.
- K. A. Shah, V. D. Gupta and K. R. Stewart, J. Pharm. Sci., 1980, 69, 594 CAS.
- M. Goldberg, in Handbook of Physiology, Section 8, Renal Physiology, ed. J. Orloff and R. W. Berliner, American Physiological Society, Washington, DC, 1973, pp. 1003–1031. Search PubMed.
- J. M. Neil, A. F. Fell and G. Smith, Int. J. Pharm., 1984, 22, 105 CrossRef CAS.
- B. Beermann, E. Dalén, B. Lindström and A. Rosén, Eur. J. Clin. Pharmacol., 1975, 9, 57 CrossRef CAS.
- M. Faed, Drug. Metab. Rev., 1984, 15, 1213 CrossRef.
- A. Rachmel, G. A. Hazelton, A. L. Yergey and D. J. Liberato, Drug. Metab. Dispos., 1985, 13, 705 Search PubMed.
- T. B. Vree, E. W. J. Beneken Kolmer, E. W. Wuis and Y. A. Hekster, Pharm. Weekbl. (Sci.), 1992, 14, 325 Search PubMed.
- T. B. Vree, M. van den Biggelaar-Martea, C. P. W. G. M. Verwey-van Wissen, M. L. Vree and P. J. M. Guelen, Br. J. Clin. Pharmacol., 1993, 35, 467 CAS.
- T. B. Vree, M. van den Biggelaar-Martea, C. P. W. G .M. Verwey-van Wissen, J. B. Vree and P. J. M. Guelen, Biopharm. Drug Dispos., 1993, 14, 491 CAS.
- J. Honori, A. D. Blair and R. E. Cutler, Clin. Pharmacol. Ther., 1977, 22, 395.
- M. Homeida, C. Roberts and R. A. Branch, Clin. Pharmacol. Ther., 1977, 22, 402 CAS.
- B. Odlind and B. Beermann, Clin. Pharmacol. Ther., 1980, 27, 784 CAS.
- A. D. Blair, A. W. Forrey, B. T. Meijsen and R. E. Cutler, J. Pharm. Sci., 1975, 64, 1334 CAS.
- A. L. M. Kerremans, Y. Tan, C. A. M. Van Ginneken and F. W. J. Gribnau, J. Chromatogr., B, 1982, 229, 129 CrossRef.
- A. K. Singh, C. McArdle, B. Gordon, M. Ashraf and K. Granley, Biomed. Chromatogr., 1989, 3, 262 CAS.
- D. Farthing, T. W. B. Gehr, I. Fakhry and D. A. Sica, LC-GC, 1991, 9, 478 Search PubMed.
- M. B. Barroso, R. M. Jiménez, R. M. Alonso and E. Ortiz, J. Chromatogr., B, 1996, 675, 303 CrossRef CAS.
- H. S. Abou-Auda, M. J. Al-Yamani, A. M. Morad, S . A. Bawazir, S. Z. Khan and K. I. Al-Khamis, J. Chromatogr., B, 1998, 710, 121 CrossRef CAS.
- A. A. Nava-Ocampo, E. Y. Velázquez-Armenta, H. Reyes-Pérez, E. Ramírez-López and H. Ponce-Monter, J. Chromatogr., B, 1999, 730, 49 CrossRef CAS.
- S. Carda-Broch, J. Esteve-Romero and M. C. García-Alvarez-Coque, J. Pharm. Biomed. Anal., 2000, 23, 803 CrossRef CAS.
- J. R. Torres-Lapasió, MICHROM Software, Marcel Dekker, New York, 2000 Search PubMed.
- T. B. Vree, M. van den Biggelaar-Martea and C. P. W. G. M. Verwey-van Wissen, J. Pharm. Pharmacol., 1995, 47, 964 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |