Molecularly imprinted CEC sorbents: investigations into polymer preparation and electrolyte composition
Leif
Schweitz
*a,
Lars I.
Andersson
b and
Staffan
Nilsson
a
aTechnical Analytical Chemistry, Center for Chemistry and Chemical Engineering, Lund University, P.O. Box 124, SE–221 00 Lund, Sweden. E-mail: leif.schweitz@teknlk.lth.se; Fax: +46 46 2224525; Tel: +46 46 2228314
bDMPK & Bioanalytical Chemistry, AstraZeneca R&D Södertälje, SE–151 85 Södertälje, Sweden
First published on 21st November 2001
Abstract
The influence of the sorbent preparation protocol and separation parameters on the selectivity and chromatographic efficiency of super-porous molecularly imprinted polymer (MIP) monoliths in capillary electrochromatography (CEC) was studied. Chiral templates were employed and enantiomer separation and resolution were used as measures of imprint selectivity and column efficiency, respectively; the latter was in addition studied by chromatography of non-related aromatic structures. The polymer preparation was varied with respect to monomer composition in the pre-polymerisation mixture and also the use of single versus multiple template(s). The separation parameters investigated were type and content of organic solvent and surfactant modifier in the electrolyte. It was found that acetone and acetonitrile in buffer mixtures provided enantiomer separation of enantiomers of the template and also structural analogues; however, the degree of separation was greatly influenced by the content in the electrolyte. Three surfactants, sodium dodecylsulfate (SDS), cetyltrimethylammonium bromide (CTAB) and polyoxyethylene sorbitanmonolaurate (Tween 20), were examined as electrolyte modifiers. It was found that addition of SDS decreased and CTAB and Tween 20 increased the enantiomer separation. SDS and CTAB could be used up to 1 mM concentration whereas Tween could be used up to 90 mM concentration without causing baseline disturbances. The effects found and demonstrated strongly suggest that these parameters are to be considered during optimisation of an MIP–CEC system.
Introduction
Molecular imprinting1–3 is recognised as an easy and effective technique to prepare polymeric matrices with selective molecular recognition properties. In the analytical chemistry field, molecularly imprinted polymers (MIPs) have found applications in solid-phase extraction,4 ligand binding assays,4 sensors5,6 and chromatography,7,8 where MIP materials offer attractive properties such as pre-determined selectivity, robustness and resistance to mechanical and chemical stress.MIPs are prepared by polymerisation of functional and cross-linking monomers in the presence of a molecular template. In the pre-polymerisation mixture the template molecule and the functional monomers form complexes, which are maintained through the polymerisation reaction. After removal of the template, polymers with imprints which are complementary to the template in terms of size, shape and chemical functionality are obtained. These polymers are able to rebind selectively the template molecule or structural analogues.
Capillary electrochromatography (CEC)9–11 has during the last decade been exposed to much research since this technique shows great promise for analytical separations. CEC is considered to combine the advantages of the high separation efficiency of capillary electrophoresis and the various retention mechanisms and selectivities offered by liquid chromatography. One important aspect of CEC is column technology which has been an active area of research.12 Several types of column design have been investigated, including packed columns,13 open-tubular columns14 and monolithic columns.15 In this regard, the use of MIPs has also been investigated as selective stationary phases in CEC.16,17 The different means of enclosing MIP sorbents within fused-silica capillaries include immobilisation of conventional imprinted polymer particles using a polyacrylamide gel,18in situ MIP preparation by a dispersion polymerisation procedure19 and an in situ photo-initiated polymerisation process for preparation of monolithic, superporous MIP sorbents.20–22 The latter approach employed derivatisation of the inner silica surface with a methacrylate monomer–silane bifunctional reagent to permit covalent attachment of the polymer to the inner wall of the capillary. The same anchoring chemistry was used to prepare thin-film coatings of MIP23 and such capillaries could be used for enantiomer separations in the open-tubular CEC mode. The superporous monoliths are currently the most successful MIP columns with respect to high selectivity and resolution; however, separation efficiencies achievable with packed silica-based particles still remain to be realised. Therefore, a number of MIP preparations were synthesised by variation of the type and content of monomers in the pre-polymerisation mixture and using either single or multiple template(s). Their behaviour in terms of enantiomer separation ability and electrochromatographic performance were evaluated under different conditions. Also, some selected electrolyte modifiers, organic solvents and surfactants were studied in order to find new approaches to optimisation of MIP-based CEC. Throughout this study the MIPs were evaluated by enantiomer separation of the imprint species or by the separation of a mixture of aromatic compounds in the CEC mode (Fig. 1). The main reason for the choice of enantiomer imprinting and separation of enantiomers is the unambiguous proof that a true imprinting effect governs the separation and not any other effect of the MIP stationary phase.
 | ||
| Fig. 1 Structures of template molecules, analytes and surfactants. Atenolol (1), metoprolol (2), propranolol (3), ropivacaine (4), benzyl alcohol (5), benzaldehyde (6), toluene (7), ethylbenzene (8), sodium dodecyl sulfate (SDS) (9), cetyltrimethylammonium bromide (CTAB) (10) and polyoxyethylene sorbitan monolaurate (Tween 20) (11). | ||
Experimental
Chemicals
Trimethylolpropane trimethacrylate [1,1,1-tris(hydroxymethyl)propane trimethacrylate; TRIM] was purchased from Aldrich (Gillingham, Dorset, UK). 2,2′-Azobis(isobutyronitrile) (AIBN), (R)-, (S)- and rac-propranolol hydrochloride and (R)-, (S)- and rac-atenolol were obtained from Sigma (St. Louis, MO, USA). (S)- and rac-metoprolol, (R)- and (S)-ropivacaine were obtained from AstraZeneca. (S)-propranolol and (S)-metoprolol were converted into their free base form by extraction with ethyl acetate and saturated NaHCO3 solution and were subsequently washed once with water and evaporated. The free bases were then stored at −20 °C until use. Toluene (HPLC grade), from Labscan (Dublin, Ireland), was dried and stored over 4 Å sieves after delivery. Sodium dodecyl sulfate (SDS) was obtained from BDH Biochemical (Poole, UK), cetyltrimethylammonium bromide (CTAB) from Merck (Darmstadt, Germany) and polyoxyethylene sorbitan monolaurate (Tween 20) from Fluka (Buchs, Switzerland). All other chemicals, including methacrylic acid (MAA), methyl methacrylate (MMA), butyl methacrylate (BMA) and epoxypropyl methacrylate (EPMA), were obtained from Merck (Hohenbrunn, Germany) and were used as received.Preparation of MIP capillary columns
A fused-silica capillary with a transparent protecting polymer coating (50, 75 or 100 μm id; 375 μm od) obtained from Polymicro Technologies (Phoenix, AZ, USA) was derivatised with [(methacryloxy)propyl]trimethoxysilane, as described elsewhere.20 The capillary was cut to the desired length and a detection window was prepared by removing about 0.5 cm of the protecting polymer layer.Pre-polymerisation mixtures were prepared by mixing monomers, cross-linking monomer, template molecule and radical initiator (AIBN) in the porogen according to Table 1. The pre-polymerisation mixture was sonicated for 5 min and introduced into the capillary. The ends were then sealed with soft plastic rubber. To perform the polymerisation reaction, the capillary was cooled to −20 °C and was illuminated by a Type TL-900 UV lamp from Camag (Muttenz, Switzerland) set at 350 nm.
| Monomer/mol l−1 | ||||||
|---|---|---|---|---|---|---|
| MIPa | MAA | MMA | BMA | EPMA | TRIM | Porogen: toluene–isooctane (v/v) |
| a The MIPs were prepared in the presence of 0.030 mol l−1 (S)-propranolol. b No template. | ||||||
| A | 0.24 | 0.24 | 99 + 1 | |||
| Bb | 0.24 | 0.24 | 98 + 2 | |||
| C | 0.12 | 0.12 | 0.24 | 99 + 1 | ||
| D | 0.12 | 0.12 | 0.24 | 98 + 2 | ||
| E | 0.18 | 0.06 | 0.24 | 98 + 2 | ||
| F | 0.06 | 0.18 | 0.24 | 98 + 2 | ||
| G | 0.12 | 0.12 | 0.24 | 99 + 1 | ||
| H | 0.12 | 0.12 | 0.24 | 98 + 2 | ||
| I | 0.18 | 0.06 | 0.24 | 98 + 2 | ||
| J | 0.06 | 0.18 | 0.24 | 98 + 2 | ||
| K | 0.12 | 0.12 | 0.24 | 99 + 1 | ||
| L | 0.24 | 0.24 | 0.48 | 95 + 5 | ||
The capillary column was flushed with several column volumes of acetonitrile, methanol and/or methanol–acetic acid (9 + 1 v/v) in order to wash the imprint molecules, the unreacted monomers and the remainder of the initiator out of the capillary column. The capillary columns were then stored at room temperature until use.
Capillary electrochromatography
Capillary electrochromatographic experiments were performed on an HP3DCE system (Hewlett-Packard, Waldbronn, Germany), consisting of a diode array detector, ChemStation software for data processing and a high-pressure facility allowing pressures up to 12 bar to be delivered to one vial or to both vials simultaneously. The electrolyte was composed of an organic solvent and buffer. Buffers were prepared by mixing 5 or 25 mM phosphoric acid with either triethanolamine (TEA) and 5 or 25 mM potassium hydroxide to the desired pH (unless stated otherwise). The samples were prepared from 10 mM water solutions diluted with water to the desired concentration. All the buffer and sample solutions were prepared using water obtained from a Milli-Q purification system (Millipore, Bedford, MA, USA). Modifiers (when applied) were added to the final electrolyte. The samples and electrolytes were degassed by sonication, injections being made electrokinetically. The separation voltage was set to 5–30 kV (usually 143–857 V cm−1) and the capillary column was thermostatted to the desired temperature. When appropriate, a resolution factor, f/g, was calculated, where f/g is defined as the ratio of the distances from a line connecting the peaks to the valley between the peaks (f) and the corresponding line to the baseline (g).24 The degree of enantiomer separation was represented by a normalised separation index ΔtR/tR(1), where ΔtR is the difference in the elution times of the enantiomers at peak maximum and tR(1) is the retention time of the first-eluted enantiomer. The capacity ratio, k′, was calculated according to k′ = (ta − tEOF)/tEOF, where ta is the elution time for the analyte and tEOF is the elution time corresponding to the EOF.Results and discussion
Monomer composition of the pre-polymerisation mixture
Most MIPs are prepared using one functional monomer and one cross-linking monomer, such as MAA and TRIM, respectively, however, in some instances more than one functional monomer has been employed and reported to improve the template recognition of the MIP.25 In the present study, the MIP was modified in that a part of the functional monomer MAA was replaced with monomers likely to be randomly incorporated into the polymer matrix and expected to interact only weakly with the template. All MIPs were prepared using 50 mol% TRIM and the remaining 50 mol% could be either of MAA, the more hydrophobic MMA and BMA and the more hydrophilic EPMA or mixtures thereof (Table 1).A series of MIP columns where 50 mol% of the MAA monomer was replaced with MMA, BMA or EPMA were prepared and compared with the MIP column prepared using MAA only as monomer (Table 1, MIPs A, C, G and K). This first set of MIP columns were evaluated by separation of rac-propranolol using standard MIP–CEC conditions [electrolyte acetonitrile–25 mM triethanolaminephosphate buffer, pH 3.0 (85 + 15 v/v); separation voltage 15 kV; column temperature 45 °C]. It was observed that the resolutions on MIPs C, G and K were higher (f/g = 1) than for MIP A (f/g = 0.8). Also, the calculated plate number for the first-eluted enantiomer was higher for MIPs C, G and K (18000–19000 plates m−1) than for MIP A (5000 plates m−1). However, the plate numbers for the last-eluted enantiomer (the imprinted analyte) were the same for all MIPs (1200–1800 plates m−1). It was also noted that the elution times for the analytes on MIP columns C and G were slightly longer than that recorded on MIP A.
In order to investigate more closely the effects of the monomers MMA and BMA, a series of MIP columns were prepared (MIPs D–E and G–J). A porogen composed of toluene–isooctane (98 + 2 v/v) was chosen since the higher isooctane content would increase the superporosity of the MIP monoliths and thus increase the probability of obtaining MIP capillary columns with good flow-through characteristics from all compositions of pre-polymerisation mixtures. However, the pre-polymerisation mixtures with the highest amounts of MMA and BMA (MIP F and J) formed dense monoliths which were impermeable to flow even at high pressures (400 bar). These MIP columns were not characterised further.
Enantiomer separations of racemic propranolol on MIP columns D,E,H and I were performed using a slightly modified conditions than above (acetonitrile–buffer 80 + 20 v/v; separation voltage 15 kV; column temperature 60 °C). The effect of incorporation of (and replacement of MAA by) MMA or BMA was an increased enantiomer separation ability in terms of resolution, normalised separation index, improved plate number for the non-imprinted enantiomer and increased elution times. The resolution (f/g) of MIP D, E, H and I was 1, 0.9, 0.8 and 0.7, respectively; hence the resolution was higher for the MMA-type than for the BMA-type MIP columns. Also, the resolution was higher in the MIP columns containing 25% MAA than in those containing 37.5% MAA. Although the highest resolution, as well as normalised separation index, was observed for the MMA-containing MIPs, the plate numbers were superior in the BMA-containing MIPs. MIP D and E showed 17000–21000 plates m−1 for the non-imprinted enantiomer whereas MIP H and I showed 30000–40000 plates m−1. The same trend was observed for the imprinted enantiomer, 3000–4000 plates m−1 for MIPs D and E and 9000–10000 plates m−1 for MIPs H and I. Owing to the low pH of the electrolyte the EOF was very low. The retention (evaluated as k′) increased with increasing amount of MMA or BMA. These monomers are more hydrophobic than the carboxylic acid monomer MAA and this effect indicates a hydrophobic interaction component in the retention mechanism (see below). Since the MMA-containing MIPs show higher retention than the BMA-containing MIPs, it may be speculated that there are differences in the degree and nature of incorporation between these monomers. Since improved enantiomer separations were seen using these ‘non-interacting’ monomers, it can be argued that these may have effects on the complex formation of template and functional monomers in the pre-polymerisation mixture leading to better defined imprints and/or effects regarding the adsorption and desorption kinetics of selective and non-selective interactions during electrochromatography caused by improved polymer characteristics.
A series of columns where all MAA monomer was replaced with either MMA, BMA or EPMA failed to achieve any enantiomer separation, which indicates the necessity for a moiety that allows strong electrostatic interaction with the analyte to be able to form molecular imprints for enantiomer separations.
In order to investigate the effects of the monomer composition regarding electroosmotic flow and retention characteristics, a series of four aromatic compounds were separated. An initial study on suitable electrolyte composition, using 5 mM potassium phosphate buffer of various pH (6–8) and various amounts of acetonitrile (40–60%), was undertaken. The analytes were benzyl alcohol, benzaldehyde, toluene and ethylbenzene (Fig. 1), which were injected electrokinetically on to MIP L with an effective length of only 8 cm.
It could be concluded that the resolution decreased with increasing acetonitrile content. At 60% acetonitrile, some analytes co-eluted regardless of the pH of the buffer. The plate numbers recorded were between 15000 and 65000 plates m−1 and the separations were typically obtained in 1 min at the highest separation potential (857 V cm−1). Although the highest resolution was found using an electrolyte composed of 40% acetonitrile and 60% pH 8 buffer, the overall performance (in terms of resolution, speed and plate number) of the electrolyte composed of 50% acetonitrile and 50% pH 7 buffer was judged to be the most adequate for the subsequent studies. Although more than sufficient for the evaluation of the various MIP preparations in this present study, the efficiency (plate numbers) is not in the same range as those which have been reported previously for non-imprinted CEC separations using packed or monolithic columns.9,15 This may be caused by the composition of the monolith. Charge density, determined by ionisable functionalities, as well as other functionalities of the monolith, can affect the EOF and also analyte interactions. Also, porosity and morphology are important. It should be remembered, however, that these monoliths are molecularly imprinted and thus primarily optimised for that.
MIP columns of different monomer compositions (MIPs D, E, H, I and B) were then evaluated using the same mixture of compounds as above and the results are summarised in Table 2 and Fig. 2. The mobility of the electroosmotic flow was calculated where the EOF was identified as a dip in the baseline originating from the water or determined by separate injections of water. It was noted that the EOF was higher in MIP columns containing a high percentage of carboxylic acid residues (Table 2). This may be explained by an increased negative charge density on the monolith which in turn promotes a higher EOF. For all analytes it is evident that a decreased MAA monomer content (and thus an increased MMA or BMA monomer content) in the pre-polymerisation mixtures results in a higher k′ value (Table 2 and Fig. 2). These k′ values may be considered as a measure of the hydrophobicity of the MIP monolith and hence the MMA and BMA MIP monoliths can be considered as being more hydrophobic in nature. Support for the contention that the retention is essentially hydrophobic driven is the fact that the k′ values are proportional to the octanol–water partition coefficients for these analytes. A higher k′ was observed on MIP D and H than on MIP E and I, respectively. The lowest k′ values were obtained for MIP B, which consisted only of MAA monomer (and cross-linker). In terms of hydrophobicity, the MIP monoliths incorporating the methyl ester monomer MMA (MIP D and E) are expected to be less hydrophobic than those incorporating the butyl ester monomer BMA (MIP H and I). The k′ values recorded were, however, higher for the MMA-containing than for the BMA-containing MIP-monoliths. Similarly, the EOF is higher for the MMA-containing than for the BMA-containing MIPs. This may be due to differences in the incorporation of the monomers [carboxylic acid monomer (MAA), ester monomers (MMA or BMA), respectively and cross-linker (TRIM)] during the polymerisation reaction. Also, differences in morphology may exist which can influence the EOF and the retention behaviour. Indicated by differences in column back-pressure and also visually, it can be assumed that a change in monomer type and composition changes the overall polymerisation system, resulting in differences in polymer morphology.
| MIP | Molar ratio: MAA∶monomer (%)a | μ EOF/m2 V−1 s−1× 10−8 | k′benzyl alcohol | k′benzaldehyde | k′toluene | k′ethylbenzene |
|---|---|---|---|---|---|---|
| a Calculated from the molar amounts in the pre-polymerisation mixture according to [MAA/(MAA + MMA + BMA + EPMA + TRIM)] × 100. | ||||||
| D | 25 | 2.76 | 0.22 | 0.40 | 0.84 | 1.1 |
| E | 37.5 | 3.25 | 0.20 | 0.34 | 0.67 | 0.87 |
| H | 25 | 2.54 | 0.17 | 0.30 | 0.62 | 0.81 |
| I | 37.5 | 2.82 | 0.15 | 0.26 | 0.51 | 0.67 |
| B | 50 | 3.68 | 0.15 | 0.23 | 0.43 | 0.54 |
 | ||
| Fig. 2 CEC separations of four aromatic compounds, benzyl alcohol, benzaldehyde, toluene and ethylbenzene (in order of elution), on capillary columns (Lt = 35 cm, Leff = 26.5 cm, id = 75 μm) of different MIP types (Table 1): (A) MIP I, (B) MIP H, (C) MIP E and (D) MIP D. Injections were made electrokinetically at 5 kV, 3 s. Separations were performed at 20 kV, 20 °C and an overpressure of 3 bar. The electrolyte was composed of acetonitrile–5 mM potassium phosphate buffer (pH 7.0) (50 + 50 v/v). Detection was made at 215 nm with a bandwidth of 10 nm. | ||
Type and amount of template used in the pre-polymerisation mixture
It has been shown previously that an MIP imprinted against a particular enantiomer is able to resolve the enantiomers of chemical analogues to that molecule in the CEC mode.20,21,26 In an attempt to broaden the applicability of MIPs in CEC, two structural analogues, (S)-atenolol and (S)-metoprolol, were used simultaneously as templates for the preparation of MIP columns (MIP O) or separately (MIPs M and N). When analysing their ability to separate the enantiomers of metoprolol and atenolol (Table 3), it was noted that enantiomer separation could be achieved on all columns. The selectivities, determined by the normalised separation index, were almost the same for atenolol and metoprolol on the two single templated columns (MIPs M and N). For both columns the resolution, however, was better for atenolol than for metoprolol, also on the (S)-metoprolol templated column. This indicates an analyte-dependent difference in both selective and non-selective interactions with the MIP. It should be considered, however, that the recognition process in CEC may be different from that during the preparation of the MIP since the environment (i.e., electrolyte and solvent) is different. For both compounds the normalised separation index was increased on the multiple templated column (MIP O) as compared with the respective single templated columns; for atenolol, the enantiomer separation was more than doubled. The underlying reasons are not fully understood, but this qualitative change is interesting and worth further study. It may be speculated that template–template interactions may play a role both in the imprinting process and during electrochromatography.27| MIP | Template (mol l−1) | Analyte (racemate) | f/g | t R1/min | t R2/min | (tR2 − tR1)/tR1 |
|---|---|---|---|---|---|---|
| a Electrolyte: acetonitrile–25 mM triethanolamine phosphate buffer (pH 3.0) (85 + 15 v/v). Electrokinetic injection at 3 kV for 7 s, separation at 5 kV and 60 °C, capillary Ltot = 35 cm, Leff = 26.5 cm, 75 μm id. Samples were of 100 μM concentration. b Columns prepared according to MIP A but with varying types of template(s). c Mixture of compounds (50 μM respectively). d Refers to the metoprolol. e Refers to the atenolol. | ||||||
| M | (S)-Metoprolol (0.030) | Metoprolol | 0.90 | 9.9 | 10.5 | 0.06 |
| Atenolol | 1 | 10.6 | 11.4 | 0.08 | ||
| N | (S)-Atenolol (0.030) | Metoprolol | 0.81 | 7.1 | 7.6 | 0.07 |
| Atenolol | 1 | 9.6 | 10.3 | 0.07 | ||
| O | (S)-Metoprolol (0.015) | Metoprolol | 0.95 | 10.3 | 11.3 | 0.10 |
| + (S)-atenolol (0.015) | Atenolol | 1 | 12.8 | 14.9 | 0.16 | |
| Metoprolol + atenololc | 0.93d | 10.3d | 11.2d | 0.09d | ||
| 1e | 13.1e | 15.4e | 0.18e | |||
Previously, conventional MIPs each imprinted with a single template have been combined and packed into a single LC column and the chromatographic results indicate that ligand–polymer cross-reactivity can be utilised to enhance certain separations.28,29 Also, the use of multiple templates with no or little structural resemblance during the synthesis of the MIP has been reported.30 The same was demonstrated here by the simultaneous imprinting of the amino alcohol (S)-atenolol and the cyclic tertiary amine (S)-ropivacaine (Fig. 1). The resultant MIP was able to resolve the enantiomers of atenolol and ropivacaine in the same CEC run which, as expected, was impossible in the single templated MIPs. Hence it is apparent that multiple templates can be used to impart imprints of more than one component in the MIP.
Modifications of the electrolyte
 | ||
| Fig. 3 Electrochromatograms of the enantiomer separation of rac-propranolol on MIP column D (Lt = 35 cm, Leff = 26.5 cm, id = 75 μm). (I) Electrolyte composed of acetone–25 mM TEA–phosphate buffer (pH 3.0) (A) 25 + 75, (B) 50 + 50 and (C) 80 + 20 v/v. Separations were performed at 15 kV, 25 °C with an applied overpressure of 3 bar. (II) Electrolyte composed of acetonitrile–25 mM TEA–phosphate buffer (pH 3.0) (A) 65 + 35, (B) 75 + 25 and (C) 80 + 20 v/v. Separations were performed at 15 kV, 60 °C with an applied overpressure of 3 bar. In all instances injections were made electrokinetically (5 kV, 10 s) from a 50 μM sample and detection was performed at 215 nm (with a bandwidth of 10 nm). | ||
According to the literature, an increase in the critical micellar concentration (cmc) occurs on addition of acetonitrile to the solvent31–33 and, in the present case, the acetonitrile concentration was >50% in the electolyte. In pure water, the cmc for SDS, CTAB and Tween 20 is reported to be 8.3, 0.92 and 0.06 mM, respectively.34 Moreover, several factors, such as organic content, salt and temperature, alter the cmc of surfactants.34 Here, the presence of micelles was unclear, but no visual evidence of micelles in the electrolyte solutions was observed. Still, the existence of micelles cannot be rejected.
Surfactant-modified electrolytes altered the performance of the enantiomer separation of propranolol. The anionic SDS decreased the resolution and the retention times of the analytes in a concentration-dependent manner. The observed effect of the cationic CTAB surfactant was an increase in resolution and also a decrease in elution times in a concentration-dependent manner. Fig. 4 shows the effect of 1 mM SDS and 1 mM CTAB in an electrolyte of acetonitrile–25 mM potassium phosphate buffer (pH 3.0) (80 + 20) as compared with the unmodified electrolyte. Increasing the SDS or CTAB concentration even further resulted in severe baseline fluctuations. The addition of increasing concentrations of Tween 20 (0–90 mM, calculated with an assumed molecular weight of 1228 g mol−1) to the electrolyte showed a general trend of improved resolution and increased elution times for the enantiomers (Fig. 5). Also, on lowering the acetonitrile concentration in the electrolyte (in this case to 50%), the resolution of the enantiomers was lost. This is expected since at higher water contents in the electrolyte the electrostatic interactions between the imprint and the analyte are decreased. However, by adding Tween 20 to this electrolyte, some of the resolution could be restored. Thus, addition of Tween 20 to a water-rich electrolyte, which by itself does not promote enantiomer separations in MIP CEC, could be used to achieve resolution of the enantiomers (Fig. 6), indicating a possible route to the use of more water-rich electrolytes for MIP–CEC enantiomer separations and opening up novel means of optimising such systems.
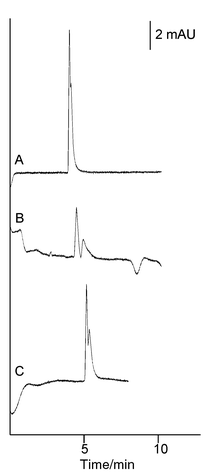 | ||
| Fig. 4 Electrochromatograms of analyses using CTAB- and SDS-modified electrolytes. Separation of rac-propranolol (25 μM) on an MIP column E (Lt = 38 cm, Leff = 29.5 cm, id = 75 μm). The electrolyte was composed of acetonitrile–25 mM TEA–phosphate buffer (pH 3.0) (80 + 20 v/v). (A) 1 mM SDS added to the electrolyte; (B) 1 mM CTAB added to the electrolyte; (C) no modifier added to the electrolyte. In all cases (S)-propranolol was the most retained analyte. Injections were made electrokinetically (3 kV, 5 s) and separations were performed at 10 kV and 60 °C. Detection at 215 nm (with a bandwidth of 10 nm). | ||
 | ||
| Fig. 5 Electrochromatograms showing the effect of Tween 20-modified electrolytes. Separation of rac-propranolol (25 μM) on an MIP column (Lt = 35 cm, Leff = 26.5 cm, id = 75 μm) type E. The electrolyte was modified with Tween 20 at concentrations of (A) 90, (B) 9, (C) 0.9 and (D) 0 mM. Conditions as in Fig. 4 with the exception that the sample was injected at 3 kV, 5 s in (C) and (D), at 5 kV, 5 s in (B) and at 15 kV, 10 s in (A) to obtain approximately the same peak height in all analyses. | ||
 | ||
| Fig. 6 Electrochromatograms of rac-propranolol analysed using (A) 90 mM Tween 20 added to the electrolyte and (B) unmodified electrolyte. The electrolyte was composed of acetonitrile–25 mM TEA–phosphate buffer (pH 3.0) (50 + 50 v/v). Injections were made at 5 kV, 5 s, otherwise all conditions were as in Fig. 5. | ||
These findings can, at least in part, be explained by the surfactants adsorbing on and modifying the polymer surface of the MIP stationary phase, which in the case of CTAB and Tween 20 are favourable for enantiomer separation. The increased elution times observed for the Tween 20-modified electrolytes may be a result of analyte interactions with possible uncharged Tween 20 micelles, although no evidence of micelles was seen. The effect can also be due to the fact that the electrophoretic mobility of the analytes is changed owing to the different electrolyte composition, and analyte interaction with free surfactant molecules may also occur. The baseline fluctuations observed at higher surfactant concentrations may be a result of micelles. The decreased elution time observed using the cationic CTAB may be a result of analyte interaction with possible micelles. Since interactions with an anionic micelle would increase the elution time, the observed effect is, however, contradictory to this proposed mechanism in the case of SDS. Again, the electrophoretic mobility is probably altered by the addition of the surfactants to the electrolyte.
Conclusions
It is concluded that the monomer content in terms of type and amount in the MIP pre-polymerisation mixture alters the characteristics of the super-porous monolithic MIP-based CEC system. Electrochromatographic behaviour such as resolution, efficiency and normalised separation index are governed by exchange of the MAA monomer in favour of MMA, BMA or EPMA. Furthermore, the use of an organic modifier (all electrolytes use an organic solvent) and/or surfactants (Tween 20 and CTAB) can improve the performance of MIP-based CEC systems. The effects found and demonstrated in this work strongly suggest that these parameters should be considered during optimisation of an MIP–CEC system. Further research into the fundamental reasons and combination effects and a deeper study of the effects presented here are warranted, although outside the scope of this investigation. We believe that these findings are valid not only for superporous monolithic MIPs in CEC, but also for MIP particle-entrapped,18 MIP coatings23,35 and MIP microparticle-based partial filling36 type CEC.Acknowledgements
We gratefully acknowledge the financial support from the Carl Trygger foundation, Kungliga Fysiografiska Sällskapet i Lund, the Swedish Natural Science Research Council (NFR), the Swedish Research Council for Engineering Sciences (TFR) and AstraZeneca.References
- K. Mosbach, Trends Biochem. Sci., 1994, 19, 9 CrossRef CAS.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812 CrossRef CAS.
- B. Sellergren, Molecularly Imprinted Polymers – Man-made Mimics of Antibodies and their Applications in Analytical Chemistry, Elsevier, Amsterdam, 1901 Search PubMed.
- L. I. Andersson, J. Chromatogr., B: Biomed. Appl., 2000, 739, 163 Search PubMed.
- F. L. Dickert and O. Hayden, Fresenius' J. Anal. Chem., 1999, 364, 506 CrossRef CAS.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495 CrossRef CAS.
- T. Takeuchi, Chromatography (Tokyo), 1999, 20, 316 Search PubMed.
- V. T. Remcho and Z. J. Tan, Anal. Chem., 1999, 71, 248A CAS.
- M. G. Cikalo, K. D. Bartle, M. M. Robson, P. Myers and M. R. Euerby, Analyst, 1998, 123, 87R RSC.
- K. D. Altria, J. Chromatogr., A, 1999, 856, 443 CrossRef CAS.
- L. A. Colon, G. Burgos, T. D. Maloney, J. M. Cintron and R. L. Rodriguez, Electrophoresis, 2000, 21, 3965 CrossRef CAS.
- Q. Tang and M. L. Lee, Trends Anal. Chem., 2000, 19, 648 CrossRef CAS.
- A. Dermaux and P. Sandra, Electrophoresis, 1999, 20, 3027 CrossRef CAS.
- K. Jinno and H. Sawada, Trends Anal. Chem., 2000, 19, 664 CrossRef CAS.
- F. Svec, E. C. Peters, D. Sykora, C. Yu and J. M. J. Frechet, J. High Resolut. Chromatogr., 2000, 23, 3 CrossRef CAS.
- P. T. Vallano and V. T. Remcho, J. Chromatogr., A, 2000, 887, 125 CrossRef CAS.
- L. Schweitz, L. I. Andersson and S. Nilsson, J. Chromatogr., A, 1998, 817, 5 CrossRef CAS.
- J. M. Lin, T. Nakagama, K. Uchiyama and T. Hobo, Chromatographia, 1996, 43, 585 Search PubMed.
- K. Nilsson, J. Lindell, O. Norrlöw and B. Sellergren, J. Chromatogr., A, 1994, 680, 57 CrossRef CAS.
- L. Schweitz, L. I. Andersson and S. Nilsson, Anal. Chem., 1997, 69, 1179 CrossRef CAS.
- L. Schweitz, L. I. Andersson and S. Nilsson, J. Chromatogr., A, 1997, 792, 401 CrossRef CAS.
- S. Nilsson, L. Schweitz and M. Petersson, Electrophoresis, 1997, 18, 884.
- Z. J. Tan and V. T. Remcho, Electrophoresis, 1998, 19, 1955.
- V. R. Meyer, Chromatographia, 1987, 24, 639 Search PubMed.
- O. Ramström, L. I. Andersson and K. Mosbach, J. Org. Chem., 1993, 58, 7562 CrossRef.
- J.-M. Lin, T. Nakagama, K. Uchiyama and T. Hobo, J. Liq. Chromatogr. Relat. Technol., 1997, 20, 1489 CAS.
- H. S. Andersson, J. G. Karlsson, S. A. Piletsky, A.-C. Koch-Schmidt, K. Mosbach and I. A. Nicholls, J. Chromatogr., A, 1999, 848, 39 CrossRef CAS.
- L. Sabourin, R. J. Ansell, K. Mosbach and I. A. Nicholls, Anal. Commun., 1998, 35, 285 RSC.
- M. A. E. Bowman, C. J. Allender, K. R. Brain and C. M. Heard, in: Drug Development Assay Approaches, Including Molecular Imprinting and Biomarkers, vol. 25, ed. E Reid, H. M. Hill and I. D. Wilson, Royal Society of Chemistry, Cambridge, 1998, p. 37 Search PubMed.
- K. Sreenivasan and R. Sivakumar, J. Appl. Polym. Sci., 1999, 71, 1823 CrossRef CAS.
- P. K. Misra, B. K. Mishra and G. B. Behera, Colloids Surf., 1991, 57, 1 CrossRef CAS.
- E. S. Ahuja and J. P. Foley, J. Chromatogr., A, 1994, 680, 73 CrossRef CAS.
- R. M. Seifar, J. C. Kraak and W. T. Kok, Anal. Chem., 1997, 69, 2772 CrossRef CAS.
- B. Jönsson, B. Lindman, K. Holmberg and B. Kronberg, Surfactants and Polymers in Aqueous Solution, Wiley, Chichester, 1998 Search PubMed.
- O. Brüggemann, R. Freitag, M. J. Whitcombe and E. N. Vulfson, J. Chromatogr., A, 1997, 781, 43 CrossRef.
- L. Schweitz, P. Spegel and S. Nilsson, Analyst, 2000, 125, 1899 RSC.
| This journal is © The Royal Society of Chemistry 2002 |