Formation and reactions of S-nitroso proteins
Darren R.
Noble
and
D. Lyn H.
Williams
*
Department of Chemistry, Durham University, South Road, Durham, UK DH1 3LE. E-mail: D.L.H.Williams@durham.ac.uk
First published on 13th December 2000
Abstract
Nitrosation of Bovine Serum Albumin (BSA) by nitrous acid occurs in two stages: a rapid step, which is consistent with an S-nitrosation reaction of a free cysteine unit, followed by a slower reaction which is consistent with an N-nitrosation reaction of a tryptophan residue. The fast reaction is catalysed by chloride and bromide ions (as is the reaction of free cysteine), whereas the slower reaction is not halide ion catalysed (like the reaction of tryptophan). Kinetic results were obtained for both reactions. The derived rate constants for the first stage for the reaction of ClNO and BrNO are reasonably close to the reported values for the reactions of cysteine. The second stage is a reversible process and we can estimate from measured infinity values and also from the variation in the measured rate constant for reactions at different [HNO2], values for the equilibrium constant of 3500 ± 200 and 2600 ± 200 dm3 mol−1 which compare reasonably with the reported value for the reaction of tryptophan of 850 dm3 mol−1. The pKa values of the cysteine residue in both BSA and Human Serum Albumin (HSA) were determined from rate measurements of the reactions with dipyridin-4-yl disulfide (4-aldrithiol), and yielded values of 8.32 and 8.18 respectively, which are close to the accepted value of 8.4 for cysteine itself, and which are substantially higher than the much quoted literature values. The S-nitroso derivatives of both BSA and HSA generated in solution (at ∼1 × 10−4 mol dm−3) showed very little sign of decomposition at pH 7.4 when measured spectrophotometrically, even in the presence of added Cu2+. There was, however, clear evidence of rapid decomposition at much lower reactant concentration (∼1 × 10−6 mol dm−3) yielding nitric oxide in increasing amounts and rates, as Cu2+ is added. These results are discussed in terms of the complexation of the Cu2+ catalyst at different albumin derivative concentrations. Transnitrosation between the S-nitroso derivatives of both BSA and HSA and excess cysteine occurred very readily. These experiments were carried out in the presence of added Cu2+, and the decomposition of the resulting S-nitrosocysteine (S-NOCys) was monitored. The results support the suggestion that S-nitroso proteins can, in theory, act as a reservoir for NO which can readily be made available, either by the Cu2+-catalysed reaction, or more readily by a direct NO+ transfer to a low molecular weight thiol, such as cysteine, which is much more prone to yield NO rapidly and quantitatively by the Cu2+-catalysed pathway. The S-nitroso derivative of BSA also underwent a relatively slow decomposition reaction initiated by cysteine, even in the presence of EDTA. This is discussed in terms of a reaction yielding nitrite anion which is not derived from NO.
The chemistry of S-nitrosothiols RSNO continues to be of much interest within the context of the formation, storage and transport of nitric oxide within the body. Nitric oxide itself, synthesised in vivo from L-arginine is known to be responsible for the control of a range of physiological functions, yet its half-life under these conditions is believed to be very short,1 too short, it is thought, for it to be transported in the body as free NO. There is a growing belief that transport and storage of NO could occur if it is bonded to thiols as RSNO species
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 2 and that NO can be obtained from them when required. There is an alternative possibility, which has yet to be convincingly demonstrated, that RSNO species (which show similar biological properties to NO) could directly transfer the NO group to an active site without the necessity for free NO formation. Whatever the answer to these questions it is important to establish the chemistry of S-nitrosothiols under conditions resembling as closely as possible those in vivo.3
2 and that NO can be obtained from them when required. There is an alternative possibility, which has yet to be convincingly demonstrated, that RSNO species (which show similar biological properties to NO) could directly transfer the NO group to an active site without the necessity for free NO formation. Whatever the answer to these questions it is important to establish the chemistry of S-nitrosothiols under conditions resembling as closely as possible those in vivo.3
The spontaneous thermal decomposition of S-nitrosothiols (often confused in the biological literature with the much faster Cu2+-catalysed reaction) is negligibly slow at room temperature, and the photochemical breakdown requires incident radiation at the appropriate wavelength. In aqueous solution RSNO compounds react at physiological pH to give nitric oxide and the disulfide RSSR4 in a process catalysed by even trace impurity levels of Cu2+. Here the true reagent5 is Cu+, generated by reduction of Cu2+ by low levels of thiolate ion (also catalytic) always present in S-nitrosothiol solutions, 6 or by any other added reducing agent, e.g. ascorbic acid7 [eqns. (1)–(3)]. Reaction can be effectively completely halted by removal of Cu2+ with EDTA, or alternatively by taking steps to reduce the thiolate concentration to trivial levels, by generating RSNO in solution with a large excess of nitrous acid over thiol.
| RSNO + H2O ⇌ RSH + HNO2 | (1) |
| 2Cu2+ + 2RS− → 2Cu+ + RSSR | (2) |
| RSNO + Cu+ → RS− + Cu2+ + NO | (3) |
Another important reaction of S-nitrosothiols in the context of possible NO formation is that wherein the NO group is transferred directly in the NO+ sense to a nucleophilic centre without generating a free NO entity. We have described a range of such reactions to nitrogen,8 sulfur9 and oxygen7,10 nucleophilic centres. This transnitrosation to a different thiolate group11,12 is of particular interest, since by this means a new R′SNO is generated [eqn. (4)] which might be much more susceptible to decomposition to give NO by the Cu2+-catalysed route than is RSNO itself. The equilibrium reaction can be driven to the right with a large excess of the thiol R′SH. It has been shown from measurements at different pH values that reaction occurs via the thiolate anion, and the mechanism shows all the characteristics of a nucleophilic reaction by thiolate anion at the nitroso nitrogen atom.
| RSNO + R′S− ⇌ RS− + R′SNO | (4) |
Another decomposition reaction of S-nitrosothiols occurs at relatively high thiol concentration. This has been studied as reactions of RSNO with RSH, i.e. with the same R group, to avoid complications due to transnitrosation. The disulfide is formed together with ammonia, nitrous oxide and nitrite ion (which is not derived from nitric oxide by oxidation and subsequent hydrolysis). This reaction is quite independent of Cu2+ and takes place in the presence of metal ion chelators. The pH–rate profile shows that the reactive entity is the thiolate ion. Reaction pathways have been proposed that account for the range of products, but these have not yet been conclusively demonstrated.13,14
In the main, all of these reactions of RSNO compounds have been studied and mechanisms established for simple low molecular weight substrate S-nitroso species, such as those of many cysteine derivatives and glutathione. In recent times a number of S-nitroso compounds including S-nitroso proteins have been detected in vivo15,16 and an S-nitroso protein derivative of serum albumin has been isolated and characterised.17,18 Since NO itself has a very short half-life in vivo, it has been proposed that NO circulates in mammalian plasma mainly as the S-nitroso derivative of serum albumin.15,19
It is clearly important to establish if S-nitroso proteins can undergo the reactions already established for simple low molecular weight RSNO structures. A number of observations have already been made, e.g. that in solution at pH 7.4, S-nitroso proteins are apparently much more resistant to breakdown to give NO than is S-nitrosocysteine itself17,20 and a number of transnitrosation reactions have been reported21 from S-nitrosocysteine (S-NOCys) and other nitrosothiols to the –SH group in proteins.
We chose to work, as others have done, with the proteins bovine serum albumin (BSA) and human serum albumin (HSA), which are reasonably plentiful in circulating plasma, are readily available and which possess a single free –SH group as Cys-34.22
Results and discussion
Thiol analysis and pKa determination of BSA and HSA
Most preparations of serum albumins contain a mixture of the one free thiol group (Cys-34) and mixed disulfides derived from it. To establish how much free thiol is present we used the analytical procedure using Ellman’s reagent,23 which develops a yellow species with a maximum absorbance at 412 nm. Our commercial samples were found to contain 57% (BSA) and 26% (HSA) free thiol respectively. These figures changed a little with each new batch of protein, and were redetermined each time.The pKa values of the free thiol groups in serum albumins have been the subject of some curiosity and uncertainty. A review article24 on the structure of serum albumins quotes an unusually low value of ∼5 for the –SH ionisation, whilst the source of the determination25 actually derives a value of about 7, which is much closer to the literature value26 of 8.4 for cysteine itself. The ‘much lower’ pKa value for the protein-bound thiol group is much quoted in the biological literature, although it has never been satisfactorily explained. We decided to redetermine these values, given the uncertainty and unusually low values quoted. We adopted the same procedure as previously25 of determining the rate constants of the reaction between the albumin and a disulfide (4-aldrithiol or dipyridin-4-yl disulfide) as a function of the pH of the solution. This reaction generates 4-mercaptopyridine which absorbs at 324 nm, and measurements were made at this wavelength. Our results for BSA are shown in Fig. 1, and show the expected sigmoid shape, if reaction occurs via the thiolate anion. Mere inspection of the graph indicates that the pKa value of the thiol is in the region of 8.5. A full analysis of the data using the Scientist package27 gave a value of 8.32 ± 0.02. A similar graph was obtained for the reaction of HSA with 4-aldrithiol, and analysis yielded a pKa value of 8.18 ± 0.02. Both of these values are quite close to the value for cysteine itself, so according to our measurements and analysis, there is no need to invoke unusually low pKa values for these albumins, since they behave in a very similar fashion to free cysteine.
 | ||
| Fig. 1 Values of kobs as a function of pH in the reaction of BSA with dipyridin-4-yl disulfide. | ||
Nitrosation of BSA and HSA
Nitrosation of both albumins was examined spectrophotometrically in aqueous acidic (∼0.4 mol dm−3 HCl) solution using sodium nitrite. Within seconds the characteristic peaks at 345 and 545 nm were generated, indicative of an S-nitrosation process. However when excess nitrous acid (over the concentration of free thiol group) was used, an additional, much slower reaction occurred resulting in a further increasing absorbance in the 345 nm region, but not in the 545 nm region of the spectrum (see Fig. 2). This does not occur when the free thiol group is present in excess over nitrous acid. It has already been suggested28 that the second nitrosation occurs at the indole nitrogen atom of a tryptophan residue within the albumin. This is borne out by these results since it has been reported29 that N-nitrosotryptophan itself has an absorbance peak at ∼340 nm with an absorption coefficient of ∼12000 dm3 mol−1 cm−1, which is an order of magnitude greater than the absorption coefficient of S-nitrosothiols at this wavelength. Tryptophan derivatives have been much used as model compounds in studies of the nitrosation of peptides and proteins.30
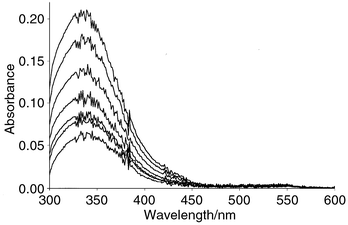 | ||
| Fig. 2 Absorbance–time plots for the nitrosation of BSA (8.8 × 10−5 mol dm−3 free thiol) with excess nitrous acid (1.83 × 10−4 mol dm−3) in HCl (0.4 mol dm−3). | ||
Kinetic measurements were made on the S-nitrosation reaction by measuring the increasing absorbance at 340 nm in a stopped-flow spectrophotometer, working as far as possible with equal concentrations of thiol function and nitrous acid. This complicated the kinetic analysis somewhat, but good fits to second-order behaviour were obtained using the Scientist package. Values of kobs [eqn. (5)] were measured as a function of added chloride and bromide ion for reactions of both BSA and HSA. The results for HSA are shown graphically in Fig. 3, where it is clear that there is halide ion catalysis, with Br− >Cl−. This is typical of S-31 and many other (but not all) nitrosation reactions. Further, from the slopes of the lines in Fig. 3 we can determine the values of the bimolecular rate constants for reaction of ClNO and BrNO with the thiol function. These are determined as 1.0 × 106 and 6.5 × 104 dm3 mol−1 s−1 for BSA and 1.1 × 106 and 13.2 × 104 dm3 mol−1 s−1 for HSA. These values are remarkably similar to the corresponding figures obtained for cysteine32 of 1.2 × 106 and 5.8 × 104 dm3 mol−1 s−1 respectively. There is a similar correspondence between the reactivity of the uncatalysed process (as measured by the value of the third-order rate constant k3 [eqn. (6)]), between S-nitrosation in the proteins and free cysteine, although these values are less precisely obtained from the intercepts of the plots in Fig. 3. These results argue strongly that we are in the present work looking at S-nitrosation of a cysteine residue within the albumins and that its reactivity is not significantly modified by its bonding within the protein.
| Rate = kobs[Thiol] [HNO2] | (5) |
| Rate = k3[RSH][HNO2][H+] | (6) |
 | ||
| Fig. 3 Catalysis by bromide ion (upper line) and chloride ion (lower line) in the S-nitrosation of HSA in HCl (0.1 mol dm−3). | ||
The second, slower reaction, when nitrous acid is in excess over the thiol, gave final absorbance values (A∞) at 340 nm which were dependent on the initial nitrous acid concentration in such a way as to suggest that the process is a reversible one, i.e.A∞ values increased with [HNO2] at constant [thiol]. Reversibility in nitrosation is well-known. It is very pronounced in the nitrosation of alcohols33 and has been identified in other cases, including the reactions of amides,34 guanidines,35 some enols,36 and N-acetyltryptophan.29 A quantitative analysis of the variation of A∞ with [HNO2], assuming a reversible process yields Fig. 4, where the points are the experimental ones, and the curve is the calculated one from the reversibility model, using the Scientist package for the fitting. This yielded a value of 3500 ± 200 dm3 mol−1 for the equilibrium constant [eqn. (7)]. Kinetic measurements also gave clear evidence of reversibility for the second nitrosation step. Measurements were carried out with a varying large excess of nitrous acid over the [thiol], and the absorbance measurements fitted to the first-order rate equation after allowance was made for the initial rapid S-nitrosation step. As is clear from the plot of the rate constants against [HNO2] (Fig. 5), there is a significant intercept to the straight line plot, characteristic of reversible reactions [eqn. (8)]. The ratio of the slope (forward rate constant k1) to the intercept (reverse rate constant k−1) gives the equilibrium constant for the second nitrosation as 2800 ± 200 dm3 mol−1, in reasonable agreement with the earlier value. These results are consistent with the suggestion that the second nitrosation occurs at the indole nitrogen atom of a tryptophan residue. The equilibrium constant for N-acetyltryptophan itself37 has been determined as 850 dm3 mol−1—reasonably close to our values. We have one further piece of evidence in support of the second nitrosation being at the tryptophan residue. The rate of the second nitrosation step in BSA carried out in 0.1 mol dm−3 HCl is totally unaffected by the presence of added sodium bromide in the range 0–0.1 mol dm−3, in sharp contrast to the catalysis noted for the faster first S-nitrosation. This is exactly what is reported for the nitrosation of free N-acetyltryptophan29,37 at pH values <1, when it is believed that proton loss from a nitrosated intermediate is the rate-limiting step.
| R′R″NH + HNO2 ⇌ R′R″NNO + H2O | (7) |
| k obs = k−1 + k1[HNO2] | (8) |
![Variation of A∞ at 340 nm with initial [HNO2] in the reaction of BSA with excess HNO2 in HCl (0.4 mol dm−3).](/image/article/2001/P2/b008112o/b008112o-f4.gif) | ||
| Fig. 4 Variation of A∞ at 340 nm with initial [HNO2] in the reaction of BSA with excess HNO2 in HCl (0.4 mol dm−3). | ||
![Plots of kobs against [HNO2] in the reaction of BSA (7.7 × 10−5 mol dm−3 free thiol) with HNO2 in HCl (0.4 mol dm−3).](/image/article/2001/P2/b008112o/b008112o-f5.gif) | ||
| Fig. 5 Plots of kobs against [HNO2] in the reaction of BSA (7.7 × 10−5 mol dm−3 free thiol) with HNO2 in HCl (0.4 mol dm−3). | ||
In summary, we have measured the pKa value of the single cysteine residues in both BSA and HSA and find values close to that of cysteine itself, contradicting the widely held belief in the biological literature that it has an unusually low value in the protein environment. The rapid S-nitrosation of the cysteine residue in BSA and HSA has been observed spectrophotometrically, and we have been able to determine rate constants for the process, which shows the normal characteristics of S-nitrosation processes of thiols, including halide ion catalysis. When nitrous acid is in excess, a second reaction occurs, which shows significant reversibility and no halide ion catalysis, and which is fully consistent with an earlier suggestion that this is an N-nitrosation process taking place at the indole nitrogen atom of a tryptophan residue in the albumin. It is known38 that BSA contains a single tryptophan unit—tryptophan 214.
Reactions of S-nitroso proteins
17,20 on the stability of these solutions. We have shown recently39 that whilst S-nitrosoglutathione (GSNO) is similarly very stable in solution at millimolar concentrations, it does break down rapidly and reasonably quantitatively to give nitric oxide at the micromolar concentration level. The explanation for this apparently very unusual result is that the disulfide product of the reaction (GSSG) binds Cu2+ very strongly, via its glutamate residues, effectively removing the catalyst. This is very much a concentration-dependent effect, and at micromolar concentrations enough Cu2+ catalyst is available. We thus examined the decomposition of S-NO BSA at much lower reactant concentrations using the NO probe to monitor the reaction. We found, as we did for GSNO, that at around and below micromolar concentrations, nitric oxide is generated, typically over 2–3 minutes. The yield of NO increased progressively from ∼10% at an initial concentration of 1.4 × 10−6 mol dm−3 to ∼60% at 3.5 × 10−8 mol dm−3. These results refer to reaction again at pH 7.4 and in the presence of added Cu2+ (1 × 10−5 mol dm−3) .We were unable to get results at lower [nitrosothiol], because the electrode system was very noisy at these low concentrations. In addition, we were not able to get reliable rate constants for NO release. We showed that this process is a Cu2+-catalysed reaction by carrying out some experiments, again with the NO probe, on the decomposition of S-NO BSA (∼1 × 10−6 mol dm−3) as a function of added Cu2+ in the range 1–5 × 10−5 mol dm−3. The results are shown in Fig. 6, where it is clear that the yield of NO increases with [Cu2+] to a maximum of 60% in this case. Even without added Cu2+ some 10% NO is generated, presumably from the residual copper present as an impurity in the buffer solution. When this is removed with EDTA, no nitric oxide is formed, confirming that in these reactions we are observing NO formation via the Cu2+-catalysed reaction.
 | ||
| Fig. 6 Yield (%) of NO in the reaction of S-NO BSA (0.92 × 10−6 mol dm−3) at pH 7.4 in the presence of (A) EDTA (1 × 10−4 mol dm−3), (B) no added EDTA or Cu2+, (C) added Cu2+ (1 × 10−5 mol dm−3), (D) added Cu2+ (3 × 10−5 mol dm−3) and (E) added Cu2+ (5 × 10−5 mol dm−3). | ||
It is now clear that although the nitrosoalbumin is very stable to decomposition in solution at millimolar concentrations, this is not the case at micromolar and lower concentrations, which is the more likely scenario in vivo. It is known40 that serum albumins will bind Cu2+ at a Gly-Gly-His peptide sequence, so it is entirely plausible that at millimolar concentrations of S-NO BSA there is effectively no free Cu2+ to bring about reaction. This effect will be much reduced as the S-NO BSA concentration is reduced to micromolar levels.
There is very little free Cu2+ in the body, but significant amounts are bound to proteins and peptides. We have previously shown41 that Cu+ can be accessed from Cu2+ bound in this way, so long as there is a reducing agent, such as a thiol or ascorbic acid present in sufficient quantity. This at least allows the possibility that NO could in principle be obtained from S-nitroso proteins in vivo.
20 that transnitrosation from an S-nitrosoalbumin is a facile process.
| S-NOAlbumin + Cys → S-NOCys → NO | (9) |
 | ||
| Fig. 7 Absorbance (at 340 nm)–time plots for the decomposition of S-NO BSA (1.5 × 10−4 mol dm−3) in the presence of Cu2+ (5 × 10−5 mol dm−3) and cysteine (Cys) in the range 2.5–15.5 × 10−3 mol dm−3. | ||
Other workers21 have noted that, as would be expected from studies on simple low molecular weight systems, S-nitrosoalbumins can be formed by transnitrosation from any RSNO to e.g. BSA. This is clearly a quite general reaction [eqn. (4)] for all R and R′ groups and the final product will depend on the relative concentrations of the reactants: the equilibrium can be driven in either direction by the use of an excess of either RSH or R′SH.
So it is clear that these S-nitrosoalbumins behave very much like the simple low molecular weight S-nitrosothiols in their ability to transfer the NO group directly to a thiolate ion, generating a new S-nitrosothiol.
We have looked briefly at one other possible S-nitroso protein decomposition reaction (which occurs also in simple RSNO species) by treatment with thiols at relatively high thiol concentration. It was found that the absorbance at 340 nm decayed in a first-order process when S-NO BSA (1.5 × 10−4 mol dm−3) was allowed to react in the presence of cysteine (1 × 10−2 mol dm−3) and of EDTA and varying concentrations of BSA (0.2–4.3 × 10−4 mol dm−3). Under these conditions it is expected that effectively all of the S-NO BSA will be converted to S-NOCys, so we are looking at the Cu2+-independent decomposition of S-NOCys brought about by BSA. The observed first-order rate constants increased linearly with [BSA]. A similar set of traces emerged when reaction occurred between S-NOCys (generated in situ) and BSA under the same conditions. We detected no nitric oxide or ammonia in the reaction products, but did find nitrite ion, although it was not formed quantitatively. These results are consistent with a rapid transnitrosation from S-NO BSA to form S-NOCys, which undergoes some reaction with BSA. The same behaviour was found for the decomposition of S-NO HSA in the presence of excess cysteine and HSA. As a check, measurements made using S-NO BSA and BSA in the presence of EDTA but no cysteine showed very little decomposition. We have not established the nature of these reactions further, but it is likely that they follow the complex course suggested by others13,14,42 for the reaction of simple RSNO species with thiols at reasonably high [thiol], in which Cu2+ is not involved. It has been suggested that nucleophilic attack by RS− occurs at the sulfur atom of RSNO leading to a variety of products, including nitrite ion and possibly involving intermediate formation of HNO.
Experimental
All materials were commercially available samples at the highest purity grade available. S-Nitrosothiols were generated in solution by conventional S-nitrosation of the corresponding thiol in acid solution, and subsequent pH adjustment. Kinetic measurements were carried out spectrophotometrically at 25 °C at pH 7.4, following the disappearance of the band at 340 nm due to RSNO in a conventional spectrophotometer interfaced with a PC, or in some cases for the faster reactions, using a stopped-flow spectrophotometer. Standard software packages were used to obtain the rate constants. Nitric oxide detection was carried out with a commercial WPI NO Mark 2 electrode calibrated using the reaction of ascorbic acid with nitrous acid. Nitrite ion was determined using the Griess analytical procedure, free thiol by the Ellman reaction and the attempted ammonia analysis using the standard diagnostic kit (Sigma).Acknowledgements
We thank the EPSRC for a Research Grant which enabled this work to be carried out.References
- M. Kelm and K. Yoshida , in Methods in Nitric Oxide Research, eds. M. Feelisch and J. S. Stamler, Wiley, 1996, pp. 49–50. Search PubMed.
- P. R. Myers, R. L. Minor, R. Guerra, J. N. Bates and D. G. Harrison, Nature, 1990, 345, 161 CAS.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869 CrossRef CAS.
- J. McAninly, D. L. H. Williams, S. C. Askew, A. R. Butler and C. Russell, J. Chem. Soc., Chem. Commun., 1993, 1758 RSC.
- A. P. Dicks, H. R. Swift, D. L. H. Williams, A. R. Butler, H. H. Al-Sa’doni and B. G. Cox, J. Chem. Soc., Perkin Trans. 2, 1996, 481 RSC.
- P. H. Beloso and D. L. H. Williams, Chem. Commun., 1997, 89 RSC.
- A. J. Holmes and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 2000, 1639 RSC.
- A. P. Munro and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1999, 1989 RSC.
- A. P. Munro and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 2000, 1794 RSC.
- P. J. Coupe and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1999, 1057 RSC.
- D. J. Meyer, H. Kramer, N. Ozer, B. Coles and B. Ketterer, FEBS Lett., 1994, 345, 177 CrossRef CAS.
- D. J. Barnett, A. Rios and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1995, 1279 RSC.
- S. P. Singh, J. S. Wishnock, M. Keshive, W. M. Deen and S. R. Tannenbaum, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14428 CrossRef CAS.
- A. P. Dicks, E. Li, A. P. Munro, H. R. Swift and D. L. H. Williams, Can. J. Chem., 1998, 76, 789 CrossRef CAS.
- J. S. Stamler, O. Jaraki, J. Osborne, D. I. Simon, J. Keaney, J. Vita, D. Singel, C. R. Valeri and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7674 CAS.
- B. Gaston, J. Reilly, J. M. Drazen, J. Fackler, P. Ramdev, D. Arnelle, M. Mullins, D. J. Sugarbaker, C. Chee, D. J. Singel, J. Loscalzo and J. S. Stamler, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10957 CAS.
- J. S. Stamler, D. I. Simon, J. A. Osborne, M. E. Mullins, O. Jaraki, T. Michel, D. J. Singel and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 444 CAS.
- D. Tsikas, J. Sandmann, S. Rossa, F.-M. Gatzki and J. C. Frolich, J. Chromatogr., B: Biomed. Appl., 1999, 726, 1 Search PubMed.
- R. M. Clancy, D. Levartovsky, J. Leszczynska-Piziak, J. Yegudin and S. B. Abramson, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3680 CAS.
- M. P. Gordge, J. S. Hothersall, G. H. Neild and A. A. N. Dutra, Br. J. Pharmacol., 1996, 119, 533 Search PubMed.
- J.-W. Park, Biochem. Biophys. Res. Commun., 1988, 152, 916 CAS.
- R. B. Simpson and H. A. Saroff, J. Am. Chem. Soc., 1958, 80, 2129 CrossRef CAS.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Anal. Biochem., 1979, 94, 75 CrossRef CAS.
- D. C. Carter and J. X. Ho, Adv. Protein Sci., 1994, 453, 153 Search PubMed.
- A. O. Pedersen and J. Jacobsen, Eur. J. Biochem., 1980, 106, 291 CAS.
- D. D. Perrin , Dissociation Constants of Organic Bases in Aqueous Solution, Butterworths, London, 1965. Search PubMed.
- Scientist package, version 2.02, MicroMath Scientific Software, Salt Lake City, UT, USA.www.micromath.com Search PubMed.
- Y.-Y. Zhang, A.-M. Xu, M. Nomen, M. Walsh, J. F. Keaney Jr. and J. Loscalzo, J. Biol. Chem., 1996, 271, 14271 CrossRef CAS.
- T. A. Meyer, D. L. H. Williams, R. Bonnett and S. L. Ooi, J. Chem. Soc., Perkin Trans. 2, 1982, 1383 RSC.
- R. Bonnett and P. Nicolaidou, Heterocycles, 1977, 7, 637 Search PubMed.
- D. L. H. Williams , Nitrosation, Cambridge University Press, 1988, pp. 173–194. Search PubMed.
- P. A. Morris and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1988, 513 RSC.
- S. E. Aldred, D. L. H. Williams and M. Garley, J. Chem. Soc., Perkin Trans. 2, 1982, 777 RSC.
- See ref. 31, p. 102..
- P. Herves, R. G. Button and D. L. H. Williams, J. Chem. Res. (S), 1998, 474 RSC.
- P. H. Beloso, P. Roy and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1991, 17 RSC.
- A. Castro, E. Iglesias, J. R. Leis, M. E. Pena, J. V. Tato and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1986, 1165 RSC.
- File ‘lao6’ Brookhaven Database. .
- D. R. Noble, H. R. Swift and D. L. H. Williams, Chem. Commun., 1999, 2317 RSC.
- S. Lau, T. P. A. Kruck and B. Sarkar, J. Biol. Chem., 1974, 249, 5878 CAS.
- A. P. Dicks and D. L. H. Williams, Chem. Biol., 1996, 3, 655 CrossRef CAS.
- P. S.-Y. Wong, J. Hyun, J. M. Fukuto, F. N. Shirota, E. G. DeMaster, D. W. Shoeman and H. T. Nagasawa, Biochemistry, 1998, 37, 5362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |