Nucleophilic substitution reactions of benzyl- and diphenylmethyl-phosphonamidic chlorides with amines: competition between the usual SN2(P) mechanism and elimination–addition with an alkylideneoxophosphorane (phosphene) intermediate
Martin J. P.
Harger
Department of Chemistry, The University, Leicester, UK LE1 7RH
First published on 5th December 2000
Abstract
The substitution reaction of PhCH2P(O)(NMe2)Cl with Me2NH or Et2NH in CHCl3 is very sensitive to the bulk of the nucleophile (![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif)
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 200 times slower with Et2NH), affords only the product derived from Me2NH in competition experiments, and gives largely undeuterated product with Et2ND; these features are in accord with an SN2(P) mechanism. The corresponding reaction of Ph2CHP(O)(NMe2)Cl is relatively insensitive to the bulk of the nucleophile (5 times slower with Et2NH), gives some of the product derived from Et2NH in competition experiments, and gives extensively deuterated product with Et2ND; these features point to an elimination–addition (EA) mechanism with an alkylideneoxophosphorane intermediate [Ph2C
200 times slower with Et2NH), affords only the product derived from Me2NH in competition experiments, and gives largely undeuterated product with Et2ND; these features are in accord with an SN2(P) mechanism. The corresponding reaction of Ph2CHP(O)(NMe2)Cl is relatively insensitive to the bulk of the nucleophile (5 times slower with Et2NH), gives some of the product derived from Et2NH in competition experiments, and gives extensively deuterated product with Et2ND; these features point to an elimination–addition (EA) mechanism with an alkylideneoxophosphorane intermediate [Ph2C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) P(O)NMe2]. There is only a modest (13–19 fold) increase in the rate of substitution on going to ArPhCHP(O)(NMe2)Cl (Ar = 4-NO2C6H4) but with R2ND there is now very rapid H/D exchange at the α carbon atom. This suggests that the elimination stage of the EA mechanism comprises rapid reversible formation of the conjugate base followed by rate-limiting expulsion of chloride ion.
P(O)NMe2]. There is only a modest (13–19 fold) increase in the rate of substitution on going to ArPhCHP(O)(NMe2)Cl (Ar = 4-NO2C6H4) but with R2ND there is now very rapid H/D exchange at the α carbon atom. This suggests that the elimination stage of the EA mechanism comprises rapid reversible formation of the conjugate base followed by rate-limiting expulsion of chloride ion.
Nucleophilic substitution at phosphoryl (P
O) centres is important in many areas of chemistry and makes possible the phosphoryl transfer reactions on which biological systems depend.1 The mechanism is generally associative [SN2(P)], with a five-coordinate intermediate or transition state.2 An alternative dissociative pathway is available to substrates that have an acidic ligand HZ (Scheme 1; X = leaving group), involving elimination–addition and a transient three-coordinate PV intermediate.2,3 This alternative mechanism is often important when Z is oxygen,4 sulfur5 or nitrogen6 but not, it seems, when Z is just a carbon atom.7 The fluoren-9-ylphosphonamidic chloride 1 is a rare exception, undergoing substitution reactions with amines by elimination–addition with an alkylideneoxophosphorane (phosphene) intermediate 2.8 This is an extreme case, however, since the Cα–H bond in 1 will be exceptionally acidic because of the aromaticity of the fluorenyl anion. In contrast are the many quite ordinary acyl and sulfonyl substrates that produce ketenes9 or sulfenes10 in their reactions with basic reagents. Is it only in extreme cases that phosphenes are formed as intermediates in the nucleophilic substitution reactions of PO compounds? That question has prompted the present investigation.
 | ||
| Scheme 1 | ||

Results and discussion
Benzylphosphonic acid derivatives
The benzylphosphonamidic chloride 4a and its 4-nitro analogue 4b were prepared by controlled reaction of the phosphonic dichlorides 3a and 3b with Me2NH (generated in situ from Me2NH2+Cl− and Et3N) (Scheme 2). Both could be purified by crystallisation but they hydrolyse quite readily and vacuum distillation proved more satisfactory for the relatively volatile and low melting compound 4a.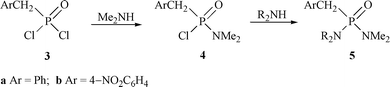 | ||
| Scheme 2 | ||
With Et2NH in CHCl3, and moisture carefully excluded, the phosphonamidic chlorides formed the phosphonic diamides 5a and 5b (R = Et) (Scheme 2) over several hours at room temperature. The reactions were monitored by 31P NMR spectroscopy (δP 46.9 or 44.0→33.9 or 32.0) using a large excess of a 2.0 mol dm−3 solution of Et2NH in CHCl3 at 30 °C and the pseudo-first-order rate constants k were deduced (Table 1).† These equate to half lives of 39 and 34 min for 4a and 4b respectively.
The corresponding reactions with Me2NH were too fast to follow so they were quenched after just 15 s (aqueous HCl) and the resulting mixtures were analysed by 31P NMR spectroscopy. In both cases the diamide product 5 (R = Me) (δP 34.2 or 33.2) accounted for 75–80%, unchanged substrate 5–10%, and by-products (most likely stemming from hydrolysis on quenching) 10–20%. This suggests half lives of ca. 7 s for the substitution reactions of 4a and 4b with Me2NH and implies at least a 200-fold difference in reactivity between Et2NH and Me2NH.
High sensitivity to the bulk of the nucleophile is not unreasonable for an SN2(P) mechanism, in which the four-coordinate phosphorus atom of the substrate becomes five-coordinate in the transition state, and it has often been noted before.11 With PhP(O)(NMe2)Cl, for example, where elimination–addition (EA) is not possible, PriNH2 reacts 120 times faster than the more bulky ButNH2,12 and with PriP(O)(NEt2)Cl the reactivity of Me2NH is 200 times greater than Et2NH.8 For an EA mechanism, however, a large steric effect would not be expected given that the amine acts as a base in the rate-limiting elimination stage, not as a nucleophile, and attacks the substrate by abstraction of a proton from the α carbon atom. Indeed, in the corresponding reactions of the benzylic PS substrate ArCH2P(S)(NMe2)Cl (Ar = 4-NO2C6H4), which are believed to proceed by elimination–addition, there is a mere 1.5 fold difference in reactivity between Et2NH and Me2NH.13
The influence of the 4-nitro substituent in the substrate 4 is also reasonable for SN2(P) but improbable for an EA mechanism. The acidity of the Cα–H bond should be greatly increased by the NO2 group and so should the rate-limiting formation of the phosphene intermediate, at least if it occurs by an ElcB mechanism. In fact the nitro-substituted substrate 4b is only marginally more reactive than 4a (Table 1). The situation with the PS substrate ArCH2P(S)(NMe2)Cl is dramatically different: a 4-nitro substituent increases the observed reactivity towards Et2NH by a factor of 2500, even though the unsubstituted compound reacts predominantly by SN2(P);13 for the EA pathway alone, the effect of the NO2 group must be to increase the rate by a factor >104.
There are two ways in which a benzylphosphonic acid derivative might become more susceptible to substitution by an EA mechanism: increased acidity of the Cα–H bond, encouraging elimination–addition, and steric hindrance of attack at the phosphorus atom, discouraging competition from SN2(P). On both counts replacing one of the benzylic hydrogen atoms by a phenyl group could have a profound effect.
Diphenylmethylphosphonic acid derivatives
Diphenylmethylphosphonic acid 8a and the related acid 8b having a 4-nitro substituent in one of the phenyl groups were obtained by hydrolysis (48% HBr; 130 °C) of the phosphonate esters 7a (R = Et) and 7b (R = Me) (Scheme 3). The first of these esters was prepared directly, by the Arbusov reaction of (EtO)3P with Ph2CHBr, but the second was obtained by an indirect route, making use of Makosza and Goliński’s vicarious substitution.14 This entailed reaction of the α-chlorobenzylphosphonate 6 (R = Me) with nitrobenzene in liquid NH3 containing NaOH. The key steps are nucleophilic attack of the phosphonate anion at the para position of nitrobenzene and, subsequently, a 1,2 shift of hydride from that position with displacement of the chlorine from the α carbon atom. | ||
| Scheme 3 | ||
The phosphonic acids were converted into the dichlorides 9 by heating with SOCl2 (DMF catalyst) and these on controlled reaction with Me2NH gave the phosphonamidic chlorides 10. The two phenyl groups in the unsubstituted compound 10a are diastereotopic, by virtue of chirality at the phosphorus atom, and that is reflected in a difference in the chemical shifts of the ortho hydrogens: δH 7.62 and 7.53 (both 2 H, d, JHH 7.5). The nitro-substituted compound 10b is chiral at carbon as well as phosphorus and was obtained as a mixture of diastereoisomers, δP 44.6 and 44.35. The NMe2 groups of the two diastereoisomers give distinct 1H NMR signals, δH 2.68 and 2.66 (both d, JPH 12.5).
The phosphonamidic chlorides 10a and 10b gave the expected diamides 12 with Me2NH and Et2NH (Scheme 4). Of these 12a (R = Me) is achiral [equivalent NMe2 groups, δH 2.40 (12 H, d, JPH 9)], 12b (R = Me) is chiral at carbon [diastereotopic NMe2 groups, δH 2.435 and 2.40 (both 6 H, d, JPH 9)], 12a (R = Et) is chiral at phosphorus [diastereotopic Ph groups; ortho protons δH 7.66 and 7.63 (both 2 H, d, JHH 8)], and 12b (R = Et) is chiral at both carbon and phosphorus and exists as diastereoisomers, δP 30.4 and 30.3 [NMe2 groups δH 2.425 and 2.39 (both d, JPH 8.5)].
 | ||
| Scheme 4 | ||
As expected the reactions of both substrates are slower with Et2NH (kE) than with Me2NH (kM) but only by a factor of about five. The difference in amine reactivity is therefore much smaller than for the benzyl compounds 4 (kM/kE
200) and much less than would be expected for SN2(P). On the other hand it is similar to the value observed with the fluorenyl substrate 1 (kM/kE = 4) which reacts with amines by an EA mechanism.8 The somewhat larger difference seen for 10a (kM/kE = 5.0) than for 10b (kM/kE = 3.6) may indicate that only the latter (nitro-substituted) substrate reacts entirely by elimination–addition. Competition from SN2(P) will be more important with the less acidic substrate 10a and with the more nucleophilic (less hindered) amine, so the observed value of kM for 10a with Me2NH may well be rather greater than the value for the EA pathway alone.
A contribution from SN2(P) in the case of Me2NH and 10a may also explain why introduction of a NO2 group into the substrate (as in 10b) has rather less impact on the observed rate of reaction with Me2NH (13-fold increase) than with Et2NH (19-fold increase). More important, however, is the fact that the NO2 group effect is so modest, regardless of the amine. It is no longer marginal, as it was for the benzyl substrate 4, but it is still nowhere near as great as for the EA pathway in the case of the PS substrate ArCH2P(S)(NMe2)Cl (>104-fold).13 That being so, an EA mechanism in which removal of the proton from the α carbon atom is rate-limiting seems untenable for the substrates 10. If they do react by an EA mechanism, the elimination stage must surely be E2, or reversible E1cB with formation of the conjugate base faster (or much faster) than its collapse (Scheme 5).
 | ||
| Scheme 5 | ||
With both Me2ND and Et2ND the nitro-substituted substrate 10b exchanged so rapidly that the integral for the methine group had fallen close to its equilibrium value [0.15–0.2 H; R2ND (large excess) contained 15–20% R2NH] by the time the first spectrum had been recorded, but still before any appreciable conversion into product had occurred. The unsubstituted compound 10a exchanges much less quickly, as expected, but still faster than its conversion into product or at least as fast. It follows that substitution could proceed by an EA mechanism in which rapid reversible formation of the conjugate base of the substrate is followed by rate-limiting expulsion of chloride ion and liberation of the phosphene intermediate (Scheme 5). It does not follow that this is the mechanism, however: unless collapse of the conjugate base is sufficiently fast the preferred route to product will still be bimolecular nucleophilic attack [SN2(P)] on the substrate itself. That, indeed, is what prevails with the nitro-substituted benzyl substrate 4b. Little if any of its substitution reaction with Et2NH proceeds by elimination–addition, yet with Et2ND we observed extensive H/D exchange in the methylene group of the substrate. With the unsubstituted benzyl substrate 4a there was not much exchange and only a small part of the product (<10%) contained any deuterium (EI MS).
While exchange between the substrate and R2ND is not necessarily indicative of an EA mechanism for substitution, incorporation of deuterium into the product in the course of the substitution process itself would surely point to a phosphene intermediate. It is significant, therefore, that in the reaction of 10a with Et2ND (80–85 atom% D) the methine group of the product was 80% deuteriated (0.2 H/0.8 D by 1H NMR) at 50% completion. At that time the methine group of the substrate was 65% deuteriated (0.35 H/0.65 D) and most of the product would have been derived from substrate that was less extensively deuteriated (exchanged). So for some of the substitution, and probably for all of it, the product must have been formed by an EA mechanism. Then a H atom could be removed in the elimination stage (phosphene formation) and a D atom acquired in the addition (phosphene + Et2ND).‡ The other substitution reactions (10a + Me2ND; 10b + Et2ND or Me2ND) also gave highly deuteriated products but in those cases H/D exchange in the substrate was so fast, relative to substitution, that we could not actually demonstrate that they were formed from less highly deuteriated starting materials.
It was not possible to compare the rates of H/D exchange with the different amines in the case of the nitro-substituted substrate 10b—they were too fast to follow—but for 10a it could be seen that exchange with Et2ND is some 20 times slower than with Me2ND (Table 2). This in spite of the fact that Et2NH is a stronger base than Me2NH. The difference in basicity is small for aqueous solutions [pKa 10.68 (Me2NH); 11.02 (Et2NH)]15 but it is quite large in the gas phase [R2NH + NH4+⇌R2NH2+ + NH3; ΔG°
−64.9 (Me2NH); −84.6 (Et2NH) kJ mol−1]16 and might be in CHCl3 solution as well.§ That being so, the relatively low kinetic basicity of Et2NH seen here must be a consequence of steric hindrance. Some steric effect is to be expected, even for abstraction of a proton, but we had supposed it would be small. In fact it seems quite large, reflecting, presumably, a crowded environment for the methine hydrogen in the Ph2CHP(O) group.
99%), in accord with an associative [SN2(P)] mechanism in which formation of the five-coordinate transition state is highly sensitive to steric effects, the diphenylmethyl substrates 10a and 10b gave substantial amounts of the products derived from Et2NH. This is easily understood in terms of an EA mechanism since the product would then arise by nucleophilic attack on a reactive and sterically accessible three-coordinate phosphene intermediate and relatively little discrimination between the amines would be expected. The NMe2∶NEt2 product ratio was actually 3.1∶1 for the nitro compound 10b but 6.4∶1 for 10a. It is possible that the phosphene intermediate from 10a reacts more selectively but much of the difference probably originates elsewhere. The EA mechanism is slower for 10a than for 10b so SN2(P) has more chance to compete, and any of the substrate that does react by SN2(P) will give entirely the product derived from Me2NH. When the experiments were repeated with a small amount of the strong base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) included in the reaction mixture (5 mol% of total amine) the NMe2∶NEt2 product ratio was practically unchanged for 10b (3.2∶1). For 10a, however, it was reduced to 3.05∶1, presumably because SN2(P) cannot now compete effectively with the DBU-assisted EA mechanism. The rate measurements hinted at some contribution from SN2(P) in the reaction of 10a with Me2NH and that is now confirmed by the outcome of the competition experiments.
Conclusion
The simple benzylphosphonamidic chloride 4a reacts with Me2NH and Et2NH by the normal associative [SN2(P)] mechanism; there is no evidence of any appreciable competition from elimination–addition, even when the acidity of the Cα–H bonds is greatly increased by the presence of a 4-nitro substituent, as in 4b. The nitro-substituted substrate is much less reactive towards Et2NH than is the corresponding PS compound [ArCH2P(S)(NMe2)Cl] for which an EA mechanism has previously been established (PO: t½ 34 min with 2.0 mol dm−3 Et2NH; PS: t½ 0.6 min with 0.8 mol dm−3 Et2NH). If the EA mechanism were as fast for the PO compound it would, in fact, be the preferred pathway, notwithstanding the relatively high SN2(P) reactivity. The Cα–H bonds may be less acidic in the PO compound,17 but most evidence suggests that any difference will be marginal18 and in any case the rapid H/D exchange seen in the reaction of 4b with Et2ND implies that the conjugate base is formed readily. Rather, it seems that elimination of chloride from the conjugate base is more difficult for the PO compound, to form a phosphene, than for the PS compound, forming a thiophosphene.
For the diphenylmethylphosphonamidic chloride 10a the reaction with Me2NH is in part elimination–addition and in part SN2(P). However, the EA mechanism is dominant with Et2NH [steric hindrance; SN2(P) suppressed] and for both amines in the case of the 4-nitro substituted substrate 10b (enhanced Cα–H acidity; EA promoted). The importance of elimination–addition here, but not with the benzyl substrates 4, is obviously a consequence of the extra phenyl group; it will increase the thermodynamic acidity of the Cα–H bond, and probably also the kinetic acidity, and it will hinder nucleophilic attack on the phosphorus atom. It is not possible to assess the effect of the extra phenyl group on acidity from our results (H/D exchange is not competitive with substitution for 4a and is too fast to measure for 4b) but its hindrance of SN2(P) is clearly very severe: even though the observed rate for 10a + Me2NH is boosted by a substantial contribution from the EA pathway it is still 104 times less than the rate of the SN2(P) reaction of 4a with Me2NH.
Experimental
Mps were determined using a Kofler hot-stage apparatus and are uncorrected. 1H NMR spectra were recorded on a Bruker ARX 250 or DPX 300 spectrometer (Me4Si internal standard; coupling constants J given in Hz) and 31P NMR spectra (1H decoupled) were recorded on the same instruments at 101.3 or 122.0 MHz (positive chemical shifts downfield from 85% H3PO4). Mass spectra were recorded in EI mode unless otherwise indicated on a Kratos Concept spectrometer. Amines were distilled from KOH and amine hydrochlorides were dried at 0.2 mmHg over P2O5. Dichloromethane was distilled from CaH2 and chloroform was dried over 5 Å molecular sieves. Light petroleum refers to the fraction bp 60–80 °C and ether to diethyl ether. The solutions used in rate studies were made up using dried materials and were stored over 3 Å molecular sieves at 4 °C for 15–24 h before use.Diphenylmethylphosphonic acid 8a
A mixture of Ph2CHBr (24.7 g, 0.10 mol) and (EtO)3P (22.6 g, 0.135 mol) was stirred vigorously under a gentle stream of nitrogen. The flask was placed in an oil bath preheated to 100 °C and the temperature was raised rapidly to ca. 165 °C. Heating was continued until all the Ph2CHBr had been consumed [2–3 h; δH 6.26 (s) replaced by 4.38 (d, JPH 25)]. The crude diethyl phosphonate7a (R = Et)19 (δP 25.2) was hydrolysed by stirring efficiently with 48% hydrobromic acid (6 equiv.) at 130 °C until 1H NMR spectroscopy showed no P–OEt signal (5 h). After cooling the solid was filtered off, washed thoroughly with water, and crystallised from aqueous MeOH to give diphenylmethylphosphonic acid8a (22.8 g, 92%), mp 229–231 °C (lit.,20 227–228 °C); m/z (−ES) 247 [(M − H)−, 100%]; δP(CDCl3–MeOH, 9∶1) 25.5; δH(CDCl3–MeOH, 9∶1; 250 MHz) 7.5–7.1 (10 H, m) and 4.39 (1 H, d, JPH 26).
4-Nitrophenyl(phenyl)methylphosphonic acid 8b
1-Chloro-1-phenylmethylphosphonic dichloride [δP(CDCl3) 40.3] was prepared from PhCHO and PCl3 (sealed tube; 200 °C for 8 h) as previously described21 and was added as a CH2Cl2 solution to MeOH (large excess) containing Et3N; evaporation of volatile material and extraction of the residue with ether afforded dimethyl 1-chloro-1-phenylmethylphosphonate226 (R = Me) (74%), δP(CDCl3) 19.7; δH(CDCl3; 90 MHz) 7.6–7.2 (5 H, m), 4.88 (1 H, d, JPH 15), 3.76 (3 H, d, JPH 11) and 3.52 (3 H, d, JPH 11). Following the published procedure14 the phosphonate 6 (R = Me) was allowed to react with nitrobenzene and NaOH in liquid NH3 to give dimethyl 4-nitrophenyl(phenyl)methylphosphonate7b (R = Me) (77%), δP(CDCl3) 25.9; δH(CDCl3; 90 MHz) 8.13 (2 H, d, JHH 9), 7.66 (2 H, dd, JPH 2, JHH 9), 7.6–7.2 (5 H, m), 4.51 (1 H, d, JPH 26), 3.51 (3 H, d, JPH 11) and 3.49 (3 H, d, JPH 11). The crude phosphonate 7b (R = Me) (6.1 g, 19.0 mmol) was hydrolysed by stirring with 48% hydrobromic acid (6 equiv.) at 130 °C until reaction was complete (3 h) (δP 24.7). The mixture was diluted with water (50 ml) and refrigerated overnight, and the precipitated solid was collected. It could not be crystallised satisfactorily (other than from water) but dissolution in moist ether and gradual addition of light petroleum (bp 40–60 °C) afforded pure 4-nitrophenyl(phenyl)methylphosphonic acid8b as the hemihydrate (4.3 g, 75%), mp 77–80 °C; m/z (−ES) 292 [(M − H)−, 100%]; δP(CDCl3) 27.0 br; δH(CDCl3; 250 MHz) 8.1 br (3 H; OH + 0.5 H2O), 8.04 (2 H, d, JHH 6), 7.41 (2 H, d, JHH 6), 7.25 (5 H, m) and 4.39 (1 H, d, JPH 25); νmax(Nujol)/cm−1 1520 and 1355 (NO2) (Found: C, 52.3; H, 4.3; N, 4.4. C13H12NO5P· 0.5H2O requires C, 51.7; H, 4.3; N, 4.6%). Prolonged drying over P2O5 at 0.2 mmHg and 50 °C gave practically anhydrous acid.
Phosphonic dichlorides
(a) Benzylphosphonic dichloride 3a and 4-nitrobenzylphosphonic dichloride 3b were prepared by literature methods.23,24(b) The phosphonic acid 8a or 8b was converted into the dichloride by stirring with SOCl2 (12 equiv.) and DMF (catalyst; 0.03 equiv.) at 100 °C (bath temp.) until the 31P NMR spectrum of the solution consisted of a single peak (δP ≈ 45) (1.5–3 h). Volatile material was evaporated and the residue was pumped at 0.2 mmHg. The following were obtained:
O).
Phosphonamidic chlorides
A solution of the phosphonic dichloride 3 or 9 (2.0 mmol) and Me2NH2+Cl− (2.4 mmol) in CH2Cl2 (5 ml) was stirred and cooled in ice while Et3N (4.0 mmol) in CH2Cl2 (5 ml) was added dropwise. In the case of 9b the phosphonic dichloride contained impurities (HCl, SOCl2) that consumed some of the amine and additional reagent was required to complete the conversion (δP 44→46). After stirring at room temperature for 0.3 h the mixture was concentrated and the residue, dissolved in fresh CH2Cl2, was washed with iced water. The solvent was evaporated and the crude product was distilled (Kugelrohr) (4a) or crystallised from CH2Cl2–light petroleum. The following were prepared:O) (Found: C, 49.4; H, 6.1; N, 6.5. C9H13ClNOP requires C, 49.7; H, 6.0; N, 6.4%).
O) (Found: C, 41.1; H, 4.7; N, 10.4. C9H12ClN2O3P requires C, 41.15; H, 4.6; N, 10.7%).
O) (Found: C, 61.2; H, 5.8; N, 4.75. C15H17ClNOP requires C, 61.3; H, 5.8; N, 4.8%).
O) (Found: C, 52.85; H, 4.7; N, 7.9. C15H16ClN2O3P requires C, 53.2; H, 4.8; N, 8.3%).
Phosphonamidic chloride rate studies
All materials and glassware were dried and moisture was excluded as completely as possible. The reaction medium was a 2.0 mol dm−3 solution of R2NH (R = Me or Et) in CHCl3 containing R2NH2+ Cl− (0.1 mol dm−3). The substrate (10–15 μmol) was dissolved in the reaction medium (240 μl) and the solution was transferred to a 4 mm NMR tube housed in a 5 mm tube containing D2O (NMR lock). Faster reactions were conducted in the probe of the NMR spectrometer at 30 °C. Slower reactions (sealed tubes) were maintained at 30 °C (block heater) and transferred to the spectrometer periodically. The reactions of 4a and 4b with Me2NH were complete within 10 min but in other cases the 31P NMR spectrum (1H decoupled) was recorded at regular intervals so that9 spectra were obtained as reaction progressed to 88% completion. For each spectrum the relative amounts of substrate and product were deduced from the integral. Minor by-products (hydrolysis) were seen in some of the Et2NH reactions but in total they amounted to ≤10%. First-order plots were linear, or practically so, and the values of k (± 6%) were deduced from the slopes of the lines (Table 1).
When reaction was complete the volatile material was evaporated and the residue was dissolved in CH2Cl2. The solution was washed with water and the product was isolated and characterised spectroscopically (see below).
The very fast reactions of 4a and 4b with Me2NH were repeated with quenching after just 15 s by addition to an excess of 1.0 mol dm−3 hydrochloric acid. The organic layer was separated, dried over anhydrous Na2CO3, and examined by 31P NMR spectroscopy. The diamide product accounted for 75–80% of the total spectrum, implying that ca. two half lives had elapsed before quenching. (Only 6–8% substrate remained but the substantial by-products were supposed to be a consequence of quenching as they were not present in reactions allowed to proceed to completion.)
Phosphonamidic chloride deuterium incorporation experiments
The salts R2ND2+Cl− (R = Me or Et) were prepared by dissolving R2NH2+Cl− in D2O (2 equiv.) and evaporating to dryness, and repeating four more times.A solution of Me2ND in CDCl3 was obtained by adding NaOD (6 mmol) in D2O (0.2 ml) to a stirred solution of Me2ND2+Cl− (0.42 g, 5 mmol) in CDCl3 (1.9 ml) at 0 °C. The organic layer was separated, shaken with solid NaCl, and dried over anhydrous K2CO3 and then over a little 3 Å molecular sieve. A sample was examined by 1H NMR spectroscopy to assess the deuterium content of the amine (NH integral 0.2 H) and its concentration (Me2N integral relative to added CH2Cl2; 1.9 mol dm−3).¶
A solution of Et2ND in CDCl3 was obtained by shaking Et2NH (1.1 g, 15 mmol) in CDCl3 (5 ml) with D2O (20 mmol) containing a little NaCl. The organic layer was collected and the process was repeated four more times. The resulting solution was dried and analysed as above (NH integral 0.15 H; amine concentration 2.5 mol dm−3 but 2.0 mol dm−3 after dilution with more CDCl3).∥
The behaviour of the phosphonamidic chlorides 4 and 10 (12–16 μmol) with R2ND (R = Me or Et) in CDCl3 (containing 0.1 mol dm−3 R2ND2+Cl−) was examined using the solutions obtained above (65 μl portions) in capillary NMR tubes. At intervals the 31P and 1H NMR spectra were recorded and from them the extent of reaction and the deuterium content of the substrate (and sometimes the product) were estimated (see Results and discussion). The temperature was generally maintained at ca. 30 °C (or 18 °C for 10b + Me2ND) but no attempt was made to obtain precise rate data. Selected results are shown in Table 2.
Phosphonamidic chloride competition experiments
The phosphonamidic chloride 4 or 10 (10–15 μmol) was added to an equimolar mixture of Me2NH and Et2NH (each 2.0 mol dm−3) in CHCl3 (containing Me2NH2+Cl− and Et2NH2+Cl−, each 0.05 mol dm−3) (150–250 μl) at ca. 30 °C. After several hours (days for 10a; reaction conducted in sealed ampoule) volatile material was evaporated and the residue was dissolved in CDCl3. The NMe2∶NEt2 product ratio was determined by 31P NMR spectroscopy. The experiments with 10a and 10b were also carried out with 1,8-diazabicyclo[5.4.0]undec-7-ene (0.2 mol dm−3) present in the reaction medium (amine hydrochloride omitted).Phosphonamidic chloride reaction products
The identities of the phosphonic diamide products 5 and 12 from the rate studies were confirmed as detailed below (crystallised from CH2Cl2–light petroleum unless indicated otherwise).From 4a and Me2NH, product5a (R = Me), mp 78–80 °C (lit.,25 79.5–80.5 °C); m/z 226 (M+, 20%) and 135 (M+ − CH2Ph, 100); δP(CDCl3) 34.2; δH(CDCl3; 250 MHz) 7.4–7.2 (5 H, m), 3.21 (2 H, d, JPH 17) and 2.54 (12 H, d, JPH 9.5); νmax(melt)/cm−1 1210 and 1190 (PO).
From 4a and Et2NH, product5a (R = Et), bp 150 °C (oven temp.) at 0.2 mmHg, solidifies at room temperature; m/z 254 (M+, 11%) and 163 (M+ − CH2Ph, 100); δP(CDCl3) 33.3; δH(CDCl3, 250 MHz) 7.4–7.15 (5 H, m), 3.18 (2 H, d, JPH 16.5), 2.97 (4 H, m), 2.54 (6 H, d, JPH 9.5) and 1.00 (6 H, t, JHH 7); νmax(melt)/cm−1 1220 (PO) (Found: M+, 254.1548; C13H23N2OP requires M, 254.1548).
From 4b and Me2NH, product5b (R = Me), mp 132–134 °C; m/z 271 (M+, 13%), 151 (14) and 135 (M+ − ArCH2, 100); δP(CDCl3) 32.3; δH(CDCl3; 250 MHz) 8.17 (2 H, d, JHH 8.5), 7.50 (2 H, dd, JPH 2, JHH 8.5), 3.30 (2 H, d, JPH 17) and 2.57 (12 H, d, JPH 10); νmax(Nujol)/cm−1 1520 and 1350 (NO2), 1210 and 1190 (PO) (Found: M+, 271.1085. C11H18N3O3P requires M, 271.1086).
From 4b and Et2NH, product5b (R = Et), mp 114–116 °C (from ether); m/z 299 (M+, 15%) and 163 (M+ − ArCH2, 100); δP(CDCl3) 31.4; δH(CDCl3; 250 MHz) 8.18 (2 H, d, JHH 8.5), 7.53 (2 H, dd, JPH 2, JHH 8.5), 3.28 (2 H, d, JPH 17), 2.97 (4 H, m), 2.58 (6 H, d, JPH 9.5) and 1.03 (6 H, t, JHH 7); νmax(Nujol)/cm−1 1515 and 1350 (NO2), 1215 and 1200 (PO) (Found: M+, 299.1398. C13H22N3O3P requires M, 299.1399).
From 10a and Me2NH, product12a (R = Me), mp 182–184 °C; m/z 302 (M+, 7%) and 135 (M+ − Ph2CH, 100); δP(CDCl3) 32.2; δH(CDCl3; 250 MHz) 7.64 (4 H, d, JHH 7), 7.35–7.15 (6 H, m), 4.345 (1 H, d, JPH 15) and 2.40 (12 H, d, JPH 9); νmax(Nujol)/cm−1 1200 and 1185 (PO) (Found: M+, 302.1548. C17H23N2OP requires M, 302.1548).
From 10a and Et2NH, product12a (R = Et), mp 152–154 °C; m/z 330 (M+, 8%) and 163 (M+ − Ph2CH, 100); δP(CDCl3) 31.7; δH(CDCl3; 250 MHz) 7.66 (2 H, d, JHH 8), 7.63 (2 H, d, JHH 8), 7.3–7.15 (6 H, m), 4.33 (1 H, d, JPH 16), 2.86 (4 H, m), 2.39 (6 H, d, JPH 9) and 0.86 (6 H, t, JHH 7); νmax(Nujol)/cm−1 1185 (PO) (Found: M+, 330.1861. C19H27N2OP requires M, 330.1861).
From 10b and Me2NH, product12b (R = Me), mp ca. 190 °C (softens at 180 °C); m/z 347 (M+, 11%), 196 (65), 166 (50), 165 (40) and 135 (M+ − ArPhCH, 100); δP(CDCl3) 31.0; δH(CDCl3; 250 MHz) 8.15 (2 H, d, JHH 9), 7.86 (2 H, d, JHH 9), 7.61 (2 H, d, JHH 7), 7.35–7.2 (3 H, m), 4.47 (1 H, d, JPH 15), 2.435 (6 H, d, JPH 9) and 2.40 (6 H, d, JPH 9); νmax(Nujol)/cm−1 1520 and 1355 (NO2), 1180 (PO) (Found: M+, 347.1398. C17H22N3O3P requires M, 347.1399).
From 10b and Et2NH, product12b (R = Et), mixture of diastereoisomers (melts 153–166 °C); m/z 375 (M+, 8%), 212 (15), 196 (55), 179 (25) and 163 (M+ − ArPhCH, 100); δP(CDCl3) 30.4 and 30.3 (ratio 1.2∶1); δH(CDCl3; 250 MHz) 8.15 (2 H, d, JHH 9), 7.88 (major) and 7.85 (total 2 H; both d, JHH 9), 7.60 and 7.59 (major) (total 2 H; both d, JHH 7), 7.35–7.2 (3 H, m), 4.46 and 4.45 (major) (total 1 H; both d, JPH 15), 2.88 (4 H, m), 2.425 and 2.39 (major) (total 6 H; both d, JPH 8.5), 0.90 (major) and 0.87 (total 6 H; both t, JHH 7); νmax(Nujol)/cm−1 1515 and 1350 (NO2), 1170 (PO) (Found: M+, 375.1712. C19H26N3O3P requires M, 375.1712). Prior to crystallisation the highfield 31P NMR diastereoisomer was in excess (ratio ca. 1∶2).
Acknowledgements
The assistance of Danny Paige and Mark Fielding is gratefully acknowledged.References
- D. E. C. Corbridge, Phosphorus: An Outline of its Chemistry, Biochemistry and Technology, Elsevier, Amsterdam, 1995; Search PubMed; Chemistry of Organophosphorus Compounds, ed. F. R. Hartley, Wiley, Chichester, 1996, vol. 4. Search PubMed.
- G. R. J. Thatcher and R. Kluger, Adv. Phys. Org. Chem., 1989, 25, 99 Search PubMed; D. M. Perreault and E. V. Anslyn, Angew. Chem., Int. Ed. Engl., 1997, 36, 433 CrossRef; A. Blasko and T. C. Bruice, Acc. Chem. Res., 1999, 32, 475 CrossRef CAS.
- Multiple Bonds and Low Coordination in Phosphorus Chemistry, ed. M. Regitz and O. J. Scherer, Thieme, Stuttgart, 1990, Section E. Search PubMed.
- For more recent work and references see: E. S. Lightcap and P. A. Frey, J. Am. Chem. Soc., 1992, 114, 9750 CrossRef CAS; J. M. Friedman, S. Freeman and J. R. Knowles, J. Am. Chem. Soc., 1988, 110, 1268 CrossRef CAS; L. D. Quin and S. Jankowski, J. Org. Chem., 1994, 59, 4402 CrossRef CAS.
- For more recent work and references see: P. M. Cullis and R. Misra, J. Am. Chem. Soc., 1991, 113, 9679 CrossRef CAS; S. P. Harnett and G. Lowe, J. Chem. Soc., Chem. Commun., 1987, 1416 RSC; I. E. Catrina and A. C. Hengge, J. Am. Chem. Soc., 1999, 121, 2156 CrossRef CAS See also L. D. Quin, P. Hermann and S. Jankowski, J. Org. Chem., 1996, 61, 3944 CrossRef CAS.
- A. F. Gerrard and N. K. Hamer, J. Chem. Soc. (B), 1968, 539 RSC; A. F. Gerrard and N. K. Hamer, J. Chem. Soc. (B), 1969, 369 RSC; S. Freeman and M. J. P. Harger, J. Chem. Soc., Perkin Trans. 2, 1988, 81 RSC; S. Freeman and M. J. P. Harger, J. Chem. Soc., Perkin Trans. 1, 1988, 2737 RSC; M. J. P. Harger, J. Chem. Soc., Perkin Trans. 2, 1991, 1057 RSC.
- G. Cevasco and S. Thea, J. Chem. Soc., Perkin Trans. 2, 1993, 1103 RSC; G. Cevasco and S. Thea, J. Chem. Soc., Perkin Trans. 2, 1994, 1103 RSC.
- M. J. P. Harger and B. T. Hurman, J. Chem. Soc., Perkin Trans. 1, 1998, 1383 RSC see also M. J. P. Harger and D. K. Jones, Chem. Commun., 1999, 339 RSC.
- F. I. Luknitskii and B. A. Vovsi, Russ. Chem. Rev., 1969, 38, 487 Search PubMed; B. R. Cho, Y. K. Kim, Y. J. Seung, J. C. Kim and S. Y. Pyun, J. Org. Chem., 2000, 65, 1239 CrossRef CAS and references cited therein..
- J. F. King, Acc. Chem. Res., 1975, 8, 10 CrossRef CAS; J. F. King, J. Y. L. Lam and S. Skonieczny, J. Am. Chem. Soc., 1992, 114, 1743 CrossRef CAS; J. F. King and R. Rathore, in The Chemistry of Sulfonic Acids, Esters and their Derivatives, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1991, ch. 17; Search PubMed; T. Kataoka and T. Iwama, Synlett, 1994, 1017 Search PubMed.
- L. Keay, J. Org. Chem., 1963, 28, 329 CAS; I. Dostrovsky and M. Halmann, J. Chem. Soc., 1953, 511 RSC; A. Kanavarioti, M. W. Stronach, R. J. Ketner and T. B. Hurley, J. Org. Chem., 1995, 60, 632 CrossRef CAS.
- S. Freeman and M. J. P. Harger, J. Chem. Soc., Perkin Trans. 1, 1987, 1399 RSC.
- M. J. P. Harger and B. T. Hurman, J. Chem. Res. (S), 1996, 490 Search PubMed.
- M. Makosza and J. Goliński, Angew. Chem., Int. Ed. Engl., 1982, 21, 451 CrossRef.
- Handbook of Chemistry and Physics, ed. D. R. Lide, 75th edn., 8–45, CRC Press, Boca Raton, 1994. Search PubMed.
- J. R. Malpass, in Comprehensive Organic Chemistry, ed. D. Barton and W. D. Ollis, Pergamon, Oxford, 1979, vol. 2, p. 34. Search PubMed.
- S. E. Denmark and C.-T. Chen, J. Am. Chem. Soc., 1995, 117, 11879 CrossRef CAS.
- M. I. Kabachnik, J. Gen. Chem. USSR (Engl. Transl.), 1994, 64, 993 Search PubMed.
- H. E. Zimmerman and R. T. Klun, Tetrahedron, 1978, 34, 1775 CrossRef CAS.
- B. A. Arbusov and N. P. Bogonoskeva, Sb. Statei Obshch. Khim., 1953, 2, 1144 ( Chem. Abstr. , 1966 , 64 , 4556c ) Search PubMed.
- M. J. P. Harger and A. Williams, J. Chem. Soc., Perkin Trans. 1, 1989, 563 RSC.
- D. Seyferth, R. S. Marmor and P. Hilbert, J. Org. Chem., 1971, 36, 1379 CrossRef.
- A. M. Kinnear and E. A. Perren, J. Chem. Soc., 1952, 3437 RSC.
- M. P. Coogan and M. J. P. Harger, J. Chem. Soc., Perkin Trans. 2, 1994, 2101 RSC.
- C. J. Cramer, S. E. Denmark, P. C. Miller, R. L. Dorow, K. A. Swiss and S. R. Wilson, J. Am. Chem. Soc., 1994, 116, 2437 CrossRef CAS.
Footnotes |
| † In substitution reactions of phosphonamidic chlorides with amines the amine hydrochloride by-product can accelerate the reaction slightly, causing deviations from linearity in the first-order rate plots (ref. 8). In the present study a small amount of the appropriate amine hydrochloride (Et2NH2Cl or Me2NH2Cl) (0.1 mol dm−3) was included in each reaction mixture and the rate plots showed little if any curvature. |
| ‡ A control reaction showed H/D exchange between the product 12a (R = Et) and Et2ND to be insignificant under the conditions of reaction (no change in 1H NMR or MS during 113 h). |
| § There is, however, only a small difference between Me2NH and Et2NH in ion-pair formation with 2,4-dinitrophenol in CHCl3 (R. G. Pearson and D. C. Vogelsong, J. Am. Chem. Soc., 1958, 80, 1038). |
| ¶ The deuterium content was not as high as expected, apparently because some D/H exchange occurred with the molecular sieve. |
| This journal is © The Royal Society of Chemistry 2001 |