The electron transfer photochemistry of allenes with cyanoarenes. Photochemical nucleophile–olefin combination, aromatic substitution (photo-NOCAS) and related reactions
Dino
Mangion
,
Donald R.
Arnold
*,
T. Stanley
Cameron
and
Katherine N.
Robertson
Department of Chemistry, Dalhousie University, Halifax, Nova Scotia, Canada B3H 4J3
First published on 11th December 2000
Abstract
The photochemical reactions of 1,2,4,5-tetracyanobenzene (1), 1,4-dicyanobenzene (2) and 1,4-dicyanonaphthalene (3) with tetramethylallene (4) and 1,1-dimethylallene (5) in acetonitrile–methanol solution have been investigated. Both 1 and 2 give 1∶1∶1 arene–allene–methanol products (7–12) in which addition of methanol occurs exclusively at the central allene carbon with aromatic substitution at the termini. The formation of these products is rationalised on the basis of a photoinduced electron transfer mechanism described as the photochemical nucleophile–olefin combination, aromatic substitution (photo-NOCAS) reaction. Cyanoarene 3 undergoes photochemical reactions initially similar to the photo-NOCAS reaction, but with addition to the arene occurring instead of substitution. The primary product (example, 22) was not isolated since it undergoes further photochemical reactions. Compound 22 and its dimethyl analogues react via a photoinduced intramolecular [2π + 2π] cycloaddition across the 3,4-double bond to give tricyclo 1∶1∶1 arene–allene–methanol adducts (14, 17 and 18) or add methanol and cyclise to bicyclo 1∶1∶2 arene–allene–methanol adducts (15, 19 and 20). The use of biphenyl (6) as a co-donor enhances the efficiency of all the photoinduced electron transfer reactions studied. In fact, removing 6 from the reaction mixture containing 3 and 5 diverts the reaction from the electron transfer pathway and instead gives an exciplex-mediated [4π + 2π] cycloadduct, 21. The mechanisms, with particular reference to the regiochemical selectivities observed in the photoreactions involving 5, are discussed.
Introduction
The photochemically induced electron transfer between cyanoaromatic electron acceptors and olefinic electron donors, such as simple aliphatic alkenes, conjugated and non-conjugated dienes, and various terpenes in the presence of a variety of nucleophiles has been thoroughly investigated in the context of the photochemical nucleophile–olefin combination, aromatic substitution (photo-NOCAS) reaction, and has provided invaluable mechanistic information regarding the behaviour of photogenerated radical ions.1–5 This reaction involves the combination of the three reagents (arene, olefin and nucleophile), generally in an anti-Markovnikov regioselective fashion (with a few exceptions). The reaction outline in Scheme 1 illustrates the mechanism utilising 2-methylpropene as the electron donor, with 1,4-dicyanobenzene and methanol as the electron acceptor and nucleophile respectively.3 The observed anti-Markovnikov regioselectivity is a direct consequence of the involvement of the olefin radical cation and can be either thermodynamically or kinetically controlled, depending on the nature of the nucleophile.4 An uncharged nucleophile such as methanol results initially in the formation of a distonic radical cation upon addition to the olefin radical cation. This distonic species is capable of equilibration with its alternative isomeric form via a bridged intermediate—in this case the regiochemistry is determined by the stability of the β-alkoxyalkyl radicals resulting from the irreversible deprotonation of the bridged distonic radical cations.4a Alternatively, utilising a negatively charged nucleophile such as fluoride anion leads to a kinetically controlled regioselectivity in which steric and polar factors dominate. Fluoride addition occurs at the less hindered site to give the more highly substituted β-fluoroalkyl radical, even when this leads to the less stable radical intermediate.4b | ||
| Scheme 1 | ||
In this study we expand our understanding of the mechanistic and synthetic aspects of the photo-NOCAS pathway by extending the reaction to aliphatic allenes as the electron donors.
The electron transfer photochemistry of these species is relatively unexplored. To the best of our knowledge, the only studies reported in the literature are an earlier report from our laboratory![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 5 and the work of Mariano.6 We have previously observed that irradiation of 1,4-dicyanobenzene and tetramethylallene in the presence of cyanide anion gave the photo-NOCAS product, 4-(4-cyanophenyl)-2,4-dimethylpent-2-ene-3-nitrile in 22% yield (Scheme 2).
5 and the work of Mariano.6 We have previously observed that irradiation of 1,4-dicyanobenzene and tetramethylallene in the presence of cyanide anion gave the photo-NOCAS product, 4-(4-cyanophenyl)-2,4-dimethylpent-2-ene-3-nitrile in 22% yield (Scheme 2).
 | ||
| Scheme 2 | ||
Mariano and co-workers conducted a more extensive study on a series of allenes utilising 2-phenyl-1-pyrrolin-1-ium perchlorate as the electron acceptor and methanol as the nucleophile. Photo-NOCAS adducts were the major products observed. Deprotonation of the allene radical cation (instead of nucleophilic trapping) followed by substitution at the pyrrolinium nucleus, and cycloaddition on the phenyl ring constituted important competing side-reactions; in the absence of a nucleophile, cycloaddition became the dominant reaction pathway.
As is typical with olefins, introduction of aryl groups on the allene inhibits substitution on the electron acceptor and leads to 1∶1 allene–nucleophile adducts. This is thought to be partly due to the increased steric bulk of the intermediate allene–nucleophile adduct radical inhibiting its addition to the electron acceptor radical anion, and, more importantly, due to the lower reduction potential of the radical which provides an efficient alternative pathway by enabling it to undergo reduction to the anion by the electron acceptor radical anion. Protonation of the anion yields what is formally an anti-Markovnikov allene–nucleophile addition product. Thus, Klett and Johnson reported that tetraphenyl- and triphenylallene undergo a photoinduced electron transfer-mediated methanol addition to give a vinyl ether, in contrast to the allyl ether obtained upon direct irradiation (Scheme 3).7 These results parallel studies from our laboratory with 1,1-diphenylethene and related systems.8
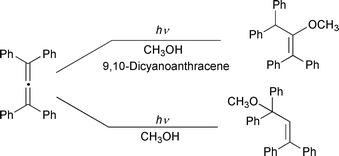 | ||
| Scheme 3 | ||
The most studied photochemical reactions of allenes are cycloadditions to ketones, cyclic enones, quinones, thiones and related systems.9–12 Although a photoinduced electron transfer mechanism has been proposed in one case,10 these reactions are usually thought to proceed via a triplet state exciplex of limited charge transfer character that collapses to a 1,4-biradical.11 However, in a more recent laser flash photolysis study on thione–allene photocycloadditions, no evidence for such an exciplex precursor was found and a mechanism involving direct formation of the biradical was suggested.12 Whatever the operating mechanism, the reaction outcome is strongly dependent both on the structure of the reactants as well as on the nature of the excited state, with [2π + 2π] cyclobutane, oxetane and various [4π + 2π] cycloadditions reported.
With a view to further defining the photochemical reactivity of allenes, we hereby present the results from our investigations into the photoinduced electron transfer reactions of the allenes, tetramethylallene (2,4-dimethylpenta-2,3-diene, 4) and 1,1-dimethylallene (3-methylbuta-1,2-diene, 5), with the cyanoarenes, 1,2,4,5-tetracyanobenzene (1), 1,4-dicyanobenzene (2) and 1,4-dicyanonaphthalene (3) in the presence of methanol as nucleophile (Fig. 1).
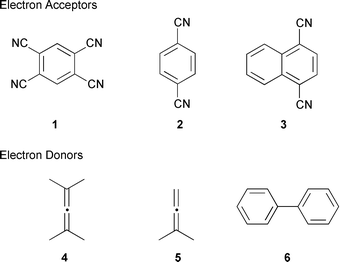 | ||
| Fig. 1 | ||
Results and discussion
Photochemistry of 1,2,4,5-tetracyanobenzene (1)
The singlet excited state of 1,2,4,5-tetracyanobenzene (1) possesses an exceptionally high reduction potential (E½red (1*) = E½red (1) + E0,0 (1) = 3.18 V, Table 1), making it a very effective electron acceptor for photoinduced electron transfer (PET). The reason why 1 has not received as much attention as an electron acceptor in PET studies as other cyanoarenes, such as 1,4-dicyanobenzene (2) and 9,10-dicyanoanthracene, is mainly due to the pronounced stability (low oxidation potential) of its radical anion. This renders it a poor photosensitiser since its radical anion is generally incapable of being oxidised back to the neutral starting material in order to complete the photosensitisation cycle.13 Nevertheless, it has proven to be very successful in PET reactions that do not require photosensitisation but rather involve the electron acceptor as a reactant. Albini and co-workers have exploited the properties of this electron acceptor in a variety of photochemical reactions with electron donors, some of which were previously inaccessible due to their relatively high oxidation potentials.14 We were thus interested in investigating how 1 would perform in a photo-NOCAS system.) and singlet excitation energies (E0,0) for the electron acceptors and half-wave oxidation potentials (E½ox) for the electron donors. All potentials are reported vs. SCE in CH3CN
| Compound |
E
½
red/Va |
E ½ ox/V |
E
0,0/eVa |
|---|---|---|---|
|
a
Ref. 15.
b
Ref. 5.
c
Peak potential Eox = 1.92 V vs. Ag/Ag+ in CH3CN, cyclic voltammetry, 100 mV s−1 scan rate, tetraethylammonium tetrafluoroborate as supporting electrolyte.16 Corrected to E½ox by subtracting 0.03 V from the peak potential;17 referenced to SCE by adding 0.34 V.
d
Ref. 1d.
|
|||
| 1,2,4,5-Tetracyanobenzene (1) | −0.65 | 3.83 | |
| 1,4-Dicyanobenzene (2) | −1.66 | 4.21 | |
| 1,4-Dicyanonaphthalene (3) | −1.28 | 3.45 | |
| Tetramethylallene (4) | +1.93b |
||
| 1,1-Dimethylallene (5) | +2.23c |
||
| Biphenyl (6) | +1.85d |
||
In our study, the electron donors have moderately high oxidation potentials (Table 1). As seen in Table 2, the free energy of the PET process involving 1 and the electron donors is highly exergonic in all cases, and electron transfer is expected to proceed at a diffusion-controlled rate.
) for the photoinduced electron transfer process between the singlet excited states of the electron acceptors and the ground state of the electron donors
| Electron acceptor | Electron donor | ΔGPET/kJ mol−1a |
|---|---|---|
|
a
Calculated using the Weller equation: ΔGPET = F(E½ox − E½red −
E0,0 − e/4πεα).18 The Coulombic attraction term Fe/4πεα was calculated as 5.4 kJ mol−1 by assuming an encounter distance α of 7 Å.1a
|
||
| 1 | 6 | −134 |
| 1 | 4 | −126 |
| 1 | 5 | −97 |
| 2 | 6 | −73 |
| 2 | 4 | −65 |
| 2 | 5 | −36 |
| 3 | 6 | −36 |
| 3 | 4 | −29 |
| 3 | 5 | 0 |
Irradiation of 1 in the presence of tetramethylallene (4) in 3∶1 acetonitrile–methanol through a Pyrex filter (λ > 280 nm) led to 24% consumption of 1 after 45 min of irradiation (Reaction 1, Table 3). The only chromatographable product formed in detectable amounts (39%) was a 1∶1∶1 arene–allene–methanol adduct, identified as 4-(2,4,5-tricyanophenyl)-3-methoxy-2,4-dimethylpent-2-ene (7) by spectroscopic methods and comparison with similar products.6b In particular, the photoproduct was distinguished from another possible isomer, 3-(2,4,5-tricyanophenyl)-4-methoxy-2,4-dimethylpent-2-ene, by consideration of the allylic C-4 resonance in the 13C NMR spectrum. The observed signal occurred at too high field (45.2 ppm) for it to be representative of an allylic carbon bearing a methoxy group.
| Reaction number | Acceptor (% consumed) | Donor | Co-donor (% recovered) | Products (% yield) |
|---|---|---|---|---|
| a In acetonitrile. b In benzene. | ||||
| 1 | 1 (24) | 4 | — | 7 (39) |
| 2 | 1 (50) | 4 | 6 (94) | 7 (48) |
| 3 | 1 (23) | 5 | — | 8 (7), 9 (1) |
| 4 | 1 (61) | 5 | 6 (98) | 8 (37), 9 (5) |
| 5 | 2 (10) | 4 | — | 10 (7) |
| 6 | 2 (46) | 4 | 6 (98) | 10 (42) |
| 7 | 2 (7) | 5 | — | 11 (21), 12 (8), 13 (8) |
| 8 | 2 (43) | 5 | 6 (95) | 11 (36), 12 (4), 13 (10) |
| 9 | 3 (10) | 4 | — | 14 (11), 16 (5) |
| 10 | 3 (75) | 4 | 6 (100) | 14 (54), 16 (24) |
| 11 | 3 (85) | 5 | — | 17 (8), 18 (22), 21 (26) |
| 12 | 3 (95) | 5 | 6 (94) | 17 (8), 18 (18), 19 (22), 20 (20) |
| 13a |
3 (84) | 5 | — | 21 (38) |
| 14b |
3 (88) | 5 | — | 21 (42) |
The inclusion of biphenyl (6) as a co-donor in the photochemical mixture had a beneficial effect on both the efficiency and yield of the photoreaction, boosting the rate of consumption to 50% after 45 min of irradiation and increasing the yield of photoproduct 7 to 48% (Reaction 2, Table 3; Scheme 4).
 | ||
| Scheme 4 | ||
The role of co-donors such as 6 is not fully understood. There are numerous reports in the literature in which PET reactions are enhanced by the addition of an appropriate co-donor, generally an aromatic hydrocarbon.19
Typically, the oxidation potential of the co-donor D is higher than that of the donor substrate Q but lower than the reduction potential of the excited state of the electron acceptor A* so that both steps shown in Scheme 5 will be exergonic. The resulting reaction enhancement is a consequence of the slower rate of back electron transfer (BET) for the A−˙–D+˙ pair as opposed to that of the A−˙–Q+˙ pair, a phenomenon attributed to a smaller reorganisation energy for D+˙ going back to D.20 Reducing the rate of BET prolongs the lifetime of the radical ion pair, allowing the two species to become efficiently solvated and to diffuse apart as free, solvated radical ions.
 | ||
| Scheme 5 | ||
However, in many cases, including the present one, the oxidation potential of the co-donor D is lower than that of the donor substrate Q, which implies that the second step in Scheme 5 is endergonic. Despite this, a marked enhancement in the photochemical reaction is commonly observed.21 The prevalent explanation put forward to account for this is that the co-donor radical cation D+˙ and the donor Q lie in equilibrium with a small amount of the donor radical cation Q+˙ and the co-donor D; consumption of Q+˙ drives the equilibrium forward generating more Q+˙. Alternatively, this may be viewed as the formation of a π-complex between D+˙ and Q that imparts substantial charge onto Q.22
Biphenyl (6) also enhances the photochemical reaction between 1,2,4,5-tetracyanobenzene (1) and 1,1-dimethylallene (5), resulting in 61% consumption of 1 after 45 min of irradiation and the formation of two isomeric 1∶1∶1 arene–allene–methanol adducts, 1-(2,4,5-tricyanophenyl)-2-methoxy-3-methylbut-2-ene (8, 37%) and 3-(2,4,5-tricyanophenyl)-2-methoxy-3-methylbut-1-ene (9, 5%) (Reactions 3 and 4, Table 3; Scheme 6). The two products are readily distinguished by the presence of two distinct resonances in both the 1H and 13C NMR spectra representing the magnetically non-equivalent methyl groups in 8; a situation not encountered in 9. Furthermore, 9 exhibits two doublets (2J = 3.6 Hz) in the 1H NMR spectrum characteristic of the two geminal olefinic protons.
 | ||
| Scheme 6 | ||
The formation of photoproducts 7, 8 and 9 can be readily explained by means of a photo-NOCAS mechanism.1–5 The mechanism, exemplified for 1,1-dimethylallene (5) as the electron donor, is illustrated in Scheme 7. Electron transfer, either directly to the excited singlet state of 1 from the ground state of the allene 5, or mediated by biphenyl (6), leads to the formation of the cyanoarene radical anion, 1−−˙, and the allene radical cation, 5++˙. The latter adds methanol, exclusively at the central carbon, to give a β-methoxyallyl radical. No products arising from addition of methanol to a terminal allenyl carbon are observed. The absence of such products is clear evidence that the allene radical cation does not add to the cyanoarene radical anion via a radical coupling mechanism while still within the geminate radical ion pair, prior to nucleophilic trapping. If this mechanism were operational we would not expect to observe the aryl moiety attached to a terminal allenyl carbon since this would generate a highly unstable vinyl cation. This alternative mode of reactivity was suggested in early studies on these types of photoreactions,23 but was largely disfavoured in subsequent work,24 with the exception of a few special cases.25 The observed reaction enhancement upon addition of biphenyl (6) is further proof against this alternative mechanism: generating the allene radical cation and the cyanoarene radical anion in separate stages should inhibit geminate radical ion pair reactions and increase the probability of interception of the allene radical cation by methanol prior to its addition onto the aromatic ring.
 | ||
| Scheme 7 | ||
The final stage in the mechanism involves the addition of the β-methoxyallyl radical to the ipso position of 1−−˙, which, as expected, is the site of highest spin density (Fig. 2). This is followed by rearomatisation of the adduct anion via expulsion of a cyanide ion to yield the final photo-NOCAS product. The unsymmetrical β-methoxyallyl radical derived from 1,1-dimethylallene (5) provides two distinct sites of reactivity. The predominance of photoproduct 8 (8∶9 = 7) in the reaction mixture indicates that the addition of the β-methoxyallyl radical to the cyanoarene radical anion is sterically controlled, with the less heavily substituted radical site being more reactive; a situation typical of radical coupling reactions.26 This observation is analogous to that reported by Mariano in which reaction at the less substituted site of the β-methoxyallyl radical of 5++˙ is favoured by a factor of 2.5 (Scheme 8).6
 | ||
| Fig. 2 Atomic charge densities with hydrogens summed into heavy atoms and total atomic spin densities (in parentheses) for the radical anions of electron acceptors 1–3 calculated by the semi-empirical AM1 method. | ||
 | ||
| Scheme 8 | ||
Photochemistry of 1,4-dicyanobenzene (2)
1,4-Dicyanobenzene (2) behaves similarly to the tetracyano analogue 1. The reduction potential of the singlet excited state (2.55 V) is somewhat lower than that of 1*; nevertheless, electron transfer with the electron donors 4–6 is expected to be highly exergonic (Table 2) and proceed at a diffusion-controlled rate. However, the photoreaction with tetramethylallene (4) is inefficient in the absence of a co-donor (Reaction 5, Table 3). A single photo-NOCAS product, identified as 4-(4-cyanophenyl)-3-methoxy-2,4-dimethylpent-2-ene (10), is obtained in low yield (7%). Addition of biphenyl (6) to the reaction mixture greatly improves the reaction, resulting in 46% consumption of 1 after 45 min of irradiation and an enhanced yield of 10 of 42% (Reaction 6, Table 3; Scheme 9).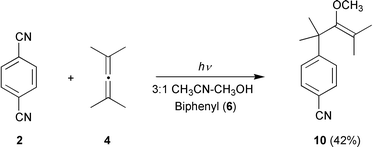 | ||
| Scheme 9 | ||
In analogy to the reaction between 1,2,4,5-tetracyanobenzene (1) and 1,1-dimethylallene (5), photolysis of 2 in the presence of 5 delivered two photo-NOCAS products, arising from reaction at the non-equivalent ambident termini of the unsymmetrical β-methoxyallyl radical derived from 5. In this case, however, the major product after 45 min of irradiation was 3-(4-cyanophenyl)-2-methoxy-3-methylbut-1-ene (11), obtained in 36% yield when biphenyl (6) was used as co-donor (Reaction 8, Table 3; Scheme 10). This is the product arising from reaction at the more sterically hindered terminus of the β-methoxyallyl radical. The isomeric 1-(4-cyanophenyl)-2-methoxy-3-methylbut-2-ene (12), which is the analogue of the major product in the similar reaction involving 1 (Reactions 3 and 4, Table 3), was detected only in trace amounts (4%). Its identity was tentatively established based solely on its GC–MS characteristics; unfortunately, we were unable to isolate this compound.
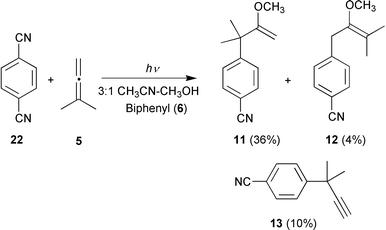 | ||
| Scheme 10 | ||
We have considered the possibility that this switch in product ratio might be due to the adventitious consumption of 12. The dimethylvinyl ether moiety is an electron-rich centre and is expected to possess a low oxidation potential (for comparison, E½ox for (Z)-2-methoxybut-2-ene is 1.73 V, SCE).27 This would enable it to undergo electron transfer with 2* in preference to 5 (E½ox = 2.23 V, SCE). Although GC monitoring of the photoreaction does show changes in the product ratio as the reaction progresses, at no point during the reaction does the product ratio favour isomer 12. The ratio of 11:12 varies from ca. 3.5 after 2 minutes to ca. 10 after 115 minutes of irradiation time. Nevertheless, the photolability of these products bearing vinyl ether moieties might account for the non-quantitative yields of the reactions.
This alteration in regiochemistry has interesting implications for the behaviour of the allylic radical intermediates. It appears that, in the absence of steric crowding at the cyanoarene reaction site (as in the case of 2), the allylic radical will react preferentially from the more highly substituted allylic terminus, which is expected to bear a greater portion of the spin density. A similar regiochemical outcome has been observed in the PET-induced substitution reaction of 2,3-dimethylbut-2-ene and 1,4-dicyanobenzene (2), which proceeds via a very similar allylic radical, generated by deprotonation of the alkene radical cation (Scheme 11).1a,f,4b,23b
 | ||
| Scheme 11 | ||
However, increasing the steric crowding at the ipso-position of the cyanoarene (as in the case of 1) causes the allylic radical to react preferentially from the less substituted terminus. The fact that, in these reactions, both isomers are present, albeit in drastically different proportions, suggests that the two regiochemical reaction pathways must differ only slightly in their activation energy barriers. Which pathway is favoured will depend on a fine balance between various contributing factors such as steric hindrance and spin density distribution. A recent study on the PET-induced addition of allylic radicals (formed via deprotonation of alkene radical cations) to phenoxy radicals (via proton abstraction by 1,4-benzoquinone radical anions) has shown similar trends as regards the regiochemical selectivity of non-symmetrical allylic radicals—in the absence of ortho-substituents on the phenoxy moiety, reaction occurred exclusively at the more highly substituted allylic terminus, but increasing the steric bulk of the ortho-substituents clearly favoured reaction at the less-substituted allylic terminus.30
The only other product evident in the chromatogram of the reaction between 2 and 5 was 3-(4-cyanophenyl)-3-methylbut-1-yne (13, 10%). This is thought to form via deprotonation of the allene radical cation 5++˙, according to the mechanism in Scheme 12. It is interesting to note that deprotonation in 5++˙ occurs exclusively from the allenyl sp2-carbon site rather than from the methyl sp3-carbon centre, a phenomenon attributed to kinetic acidity.6b,31
 | ||
| Scheme 12 | ||
Deprotonation of alkene radical cations is commonly observed as the major reaction pathway in the absence of a nucleophile. In the case of aliphatic alkenes, the allylic radical formed upon deprotonation usually substitutes, from either ambident end, at the ipso-position of the cyanoarene radical anion to give 1∶1 arene–alkene products (Scheme 11).1a,f,4b,23b With aromatic alkenes, the allylic radical does not substitute but instead gets reduced to the allylic anion, which can then get protonated at either end of the allylic moiety to regenerate the starting material or to give a deconjugated tautomer (Scheme 13).32
 | ||
| Scheme 13 | ||
Alkene radical cations are generally not acidic enough for deprotonation to compete effectively with nucleophilic trapping by strong nucleophiles such as methanol.32b Deprotonation only becomes competitive in the presence of weaker nucleophiles such as fluoride anion which also has substantial basic character.4b In contrast, the increased acidity of allene radical cations does allow deprotonation to compete with nucleophilic addition. In addition to the results obtained in this work, Mariano and co-workers also observed appreciable amounts of 1∶1 products derived from deprotonation of the allene radical cation (Scheme 8).6 Similarly, Klett and Johnson reported the formation of a deprotonation product (1,3,3-triphenylpropyne) in their PET study on triphenylallene.7b Both of these reactions were conducted in pure methanol.
Photochemistry of 1,4-dicyanonaphthalene (3)
1,4-Dicyanonaphthalene (3) possesses the lowest excited state reduction potential (2.17 V, Table 1) among the electron acceptors chosen for this investigation. This is mainly due to its low singlet excitation energy, E0,0. Nevertheless, the free energies for PET are still sufficiently exergonic to ensure an efficient electron transfer process, except in the case of 1,1-dimethylallene (5) for which the free energy is isoergic (Table 2). As in the previous experiments, biphenyl (6) was found to enhance the reactions (Table 3).Thus, the photolysis of 3 and tetramethylallene (4) in the presence of 6 gave two crystalline products upon chromatographic workup which were identified as 1,5-dicyano-3-methoxy-2,2,4,4-tetramethyl-6,7-benzotricyclo[3.2.2.03,8]nonane (14, 54%) and cis-1,4-dicyano-6,6,8,8-tetramethyl-7-oxo-2,3-benzo-cis-bicyclo[3.3.1]nonane† (16, 24%) (Reaction 10, Table 3; Scheme 14).
 | ||
| Scheme 14 | ||
The structural assignments of both products were confirmed by X-ray crystallographic analyses since spectroscopic interpretation was not trivial. In particular, the absence of vicinal coupling constants in the 1H NMR spectra of both products was quite misleading. In the case of 14, only one of the two geminal C-9 protons appeared, as expected, as a doublet of doublets (2.71 ppm, 2J = 12.8 Hz, 3J = 4.6 Hz); the other showed up as a simple doublet (1.88 ppm, 2J = 12.8 Hz) and as a consequence the vicinal C-8 proton appeared as a doublet (3.45 ppm, 3J = 4.6 Hz) as well. The Karplus equation predicts that a vicinal coupling constant is reduced to a minimum when the dihedral angle between the coupled protons is approximately 90°;33 crystallographic analysis of 14 suggested a value of 76° for one dihedral angle between the C-8 proton and one of the C-9 protons. The other dihedral angle is estimated as 47°. The observed coupling of 4.6 Hz would then represent the coupling at this 47° angle, in a situation where 0° is approximately 8 Hz. This is a reasonable value in view of the presence of electron withdrawing substituents next to the tricyclic ring protons which may contribute to a reduction in the coupling constants.33a,34
The bicyclo-ring 1H NMR resonances in 16 also displayed some peculiarities that made reliable structural elucidation difficult. One of the C-9 proton signals appeared at 3.26 ppm as a doublet of doublets of doublets, displaying a geminal coupling of 14.5 Hz to the other C-9 proton and a vicinal coupling of 4.0 Hz to the C-5 proton, as well as a long-range w-coupling of 1.5 Hz to the C-4 proton. All the couplings to the C-5 proton (2.61 ppm) were either zero or small so that its resonance appeared as a broad singlet. Furthermore, the low-field C-4 proton (4.19 ppm) was a broad apparent singlet that failed to exhibit the vicinal coupling to the C-5 proton and the w-coupling to the C-9 proton at 3.26 ppm. Compound 16 is most probably the hydrolysis product of acetal 15 (vide infra), although we were unable to isolate 15 from the reaction mixture.
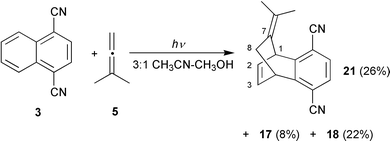
The photolysis of 3 and 1,1-dimethylallene (5) led to a more complex mixture of products due to the regiochemical possibilities arising from the involvement of the unsymmetrical allene. When biphenyl (6) was used we were able to isolate the four major products from the photochemical mixture: two 1∶1∶1 arene–allene–methanol tricyclic products, 1,5-dicyano-3-methoxy-4,4-dimethyl-6,7-benzotricyclo[3.2.2.03,8]nonane (17, 8%) and 2,5-dicyano-1-methoxy-9,9-dimethyl-3.4-benzotricyclo[3.3.1.02,7]nonane (18, 18%), and two 1∶1∶2 arene–allene–methanol bicyclic products, cis-1,4-dicyano-7,7-dimethoxy-6,6-dimethyl-2,3-benzo-cis-bicyclo[3.3.1]nonane (19, 22%) and trans-1,4-dicyano-7,7-dimethoxy-8,8-dimethyl-2,3-benzo-cis-bicyclo[3.3.1]nonane† (20, 20%) (Reaction 12, Table 3; Scheme 15).
 | ||
| Scheme 15 | ||
Spectroscopic identification of the products 17–20 was further complicated by the possibility of regioisomers. Therefore, the characterisation of these products relied mainly on their X-ray crystallographic analyses. The 1H and 13C NMR spectra of the tricyclic product 17 were very similar to those of 14. In the 13C NMR spectrum, the two lower-field methyl carbons (26.0 and 27.0 ppm) and one of the high-field quaternary carbons present in 14 were replaced by a new methylene carbon resonance at 42.1 ppm in 17. Similarly, in the 1H NMR spectrum, two methyl singlets were replaced by two coupled doublets at 2.23 and 3.12 ppm (2J = 12.2 Hz) that are representative of the two geminal methylene protons.
We were fairly confident with our assignment of the tricyclo-ring skeleton but determining the location of the gem-dimethyl group was less straightforward. The problem was resolved by means of an X-ray crystallographic structure that, despite limited observed data and relatively high R and Rw values (isotropic refinement only), provided us with sufficiently reliable structural data to identify compound 17.
Similar to the previous products, the tricyclo product 18 exhibited some unexpected coupling patterns in its 1H NMR spectrum. A 2D 1H–1H correlation NMR experiment (COSY) confirmed that the doublet of doublets of doublets at 1.71 ppm (2J = 12.8, 3J = 7.2, 4J = 1.2 Hz) and the doublet at 2.50 ppm (2J = 12.8 Hz) constituted a pair of methylene protons (at C-6 or C-8). The doublet of doublets of doublets at 2.99 ppm (2J = 10.7, 3J = 7.2, 4J = 1.2 Hz) and the doublet at 2.20 ppm (2J = 10.7 Hz) constituted the other pair of methylene protons. The pseudo-triplet at 2.62 ppm (3J = 7.2 Hz) corresponded to the bridgehead methine proton at C-7, flanked by the two methylene groups at C-6 and C-8. These assignments imply that one of the protons in either methylene group has a vicinal coupling constant of zero with the central C-7 proton. Interestingly, the other two methylene protons exhibit a long-range w-coupling (4J = 1.2 Hz) to one another. The ca. 90° dihedral angle (3J = 0) and the w-configuration of the two methylene protons (w-coupling) can be readily appreciated from a computed molecular mechanics model of the compound.
Unlike in the case of 15, we were able to isolate the acetals 19 and 20, which were found to be stable in the crystalline state. Nevertheless, GC–MS analysis of the two compounds indicated that the acetals decompose readily to the respective demethanolysis products (M+˙, m/z = 278) inside the GC injector. However, when higher concentrations of the materials were injected, a second chromatographic peak representing the acetals (M+˙, m/z = 310) largely replaced the peak due to the decomposition products. Because of this chromatographic problem, the reported yields for these species possibly underestimate the true yields.
Just as with 16, the C-4 proton signal in the 1H NMR spectrum (singlet, 4.30 ppm) of 19 did not exhibit any coupling to the C-5 proton. Both 16 and 19 have the two cyano groups arranged cis to one another. However, in acetal 20, the cyano groups are arranged in a trans geometry and the C-4 proton (4.37 ppm) appears as a doublet (3J = 6.1 Hz).
Removal of biphenyl (6) from the reaction mixture altered the course of the photochemical reaction (Reaction 11, Table 3; Scheme 16). The major product is now a 1∶1 arene–allene adduct identified as 7-isopropylidene-5,6-(1′,4′-dicyanobenzo)bicyclo[2.2.2]oct-2-ene† (21, 26%) that was absent from the reaction when 6 was used as co-donor. Also present were the tricyclo products 17 (8%) and 18 (22%) but no appreciable amounts of the acetals 19 and 20 were detected. The identity of 21 rests predominantly on an X-ray crystallographic structure analysis.
 | ||
| Scheme 16 | ||
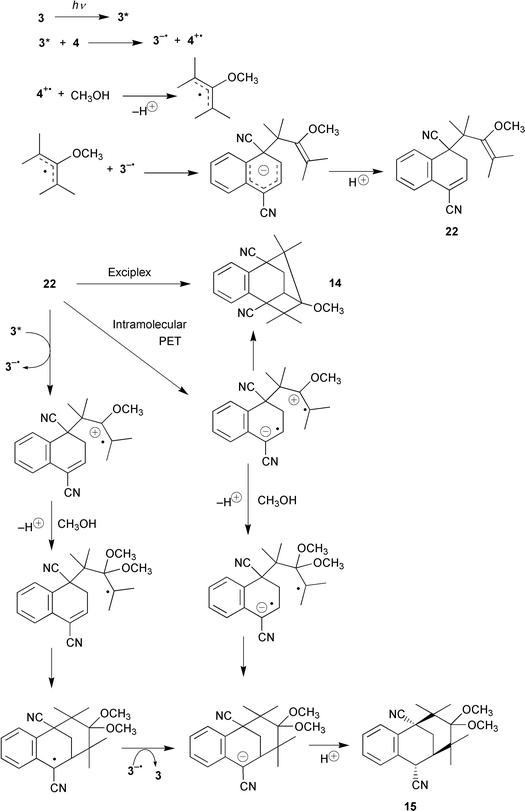
The mechanism proposed to account for the observed reactions is illustrated for the reaction between 1,4-dicyanonaphthalene (3) and tetramethylallene (4) in Scheme 17. The mechanism starts along a pathway similar to that of the photo-NOCAS reaction. The 1,4-dicyanonaphthalene radical anion, 3−−˙, and the allene radical cation, 4++˙, are photochemically generated, either directly or in separate stages involving biphenyl (6) as co-donor. (For brevity and clarity, the steps involving 6 have been omitted from Scheme 17.) The allene radical cation is intercepted by methanol to generate a β-methoxyallyl radical which adds onto an ipso-position (site of highest spin density; Fig. 2) of 3−−˙.
 | ||
| Scheme 17 | ||
At this point the mechanism diverges from the photo-NOCAS pathway. Whereas in the photo-NOCAS reaction the adduct anion rearomatises via elimination of cyanide anion, in the case of 3 the anion gets protonated to give an overall addition product, 22. This switch from substitution to addition reactions on going from a mononuclear to a binuclear aromatic system is often observed in PET reactions.23b,35 Presumably this is due to the lower rearomatisation energy gain in a binuclear as opposed to a mononuclear system.
Despite an attentive search, compound 22 could not be detected in the photochemical mixture, even at low conversions. This is not very surprising when one considers that the molecule is perfectly set up to undergo further photochemical reaction. The electron-rich vinyl ether and the electron-poor α-cyanostyryl moiety (E½red for 2-phenylpropenonitrile is −2.20 V, SCE)36 are geometrically arranged in such a way as to allow an effective charge transfer interaction. The tricyclo product 14 is formed via a [2π + 2π] cycloaddition, either directly from the exciplex or after intramolecular PET between the cyanostyryl and vinyl ether moieties. Methanol trapping of the cationic vinyl ether competes with the cycloaddition reaction. Methanol adds at the α-methoxy position that is expected to bear the higher positive charge density. The resulting acetal intermediate then cyclises via a radical–radical coupling reaction at the β-position of the cyanostyryl moiety to give an anionic adduct, which is subsequently protonated to yield the product 15. Alternatively, the radical cation of the vinyl ether moiety can be generated via an intermolecular PET with 1,4-dicyanonaphthalene (3), possibly aided by biphenyl (6). If this is the case, the radical generated upon trapping of the vinyl ether radical cation by methanol will add to the α-cyanostyryl moiety to give an α-cyanostyryl radical which should be readily reduced to the corresponding anion via electron transfer from 3−−˙. Protonation would furnish product 15. Product 15 was not isolated in the reaction involving 4, since it was hydrolysed in situ to the corresponding ketone 16. In the reaction involving 3, however, the equivalent acetals 19 and 20 survived hydrolysis and were successfully isolated.
The cis-stereochemistry in both 16 and 19 came as a surprise since this implies that protonation occurred from what appears to be the more sterically crowded side. We were not able to detect the corresponding trans isomers.
Similarly to the reaction involving 1,4-dicyanobenzene (2), the reaction between 3 and 5 yields a predominance of products arising from reaction at the more substituted terminus of the β-methoxyallyl radical derived from 5++˙ (17 + 18 + 20∶19 = 2.3). Although it is perhaps speculative to attach much significance to the absence of possible products, particularly in view of the non-quantitative yields obtained, the compounds isolated do represent the major products in the reaction.
Compounds similar to the tricycloadducts 14, 17 and 18 have been observed in a previous study from our laboratory involving the photolysis of 3 and 2,3-dimethylbut-2-ene in acetonitrile (Scheme 18).23b Under these conditions, the alkene radical cation generated upon PET deprotonates to give an allylic radical. From this stage onwards, the mechanistic pathway followed is very similar to the one leading to product 14 in Scheme 17. In this earlier study, however, the product ratio implies that addition of the allylic radical to the ipso-position of the arene radical anion occurs predominantly from the less substituted end. This contrasts with our current results and lends support to the hypothesis that the regiochemical control in these reactions is very sensitive to the controlling factors and is, therefore, hard to predict reliably.
 | ||
| Scheme 18 | ||
The effect of biphenyl (6) on the photochemical reaction between 3 and 5 merits some attention. As shown in Table 2, the free energy for the PET process between 3 and 5 is isoergic, so that electron transfer is disfavoured when no co-donor mediation is possible. The major product 21 is formally a [4π + 2π] photocycloadduct, formed via the addition of an allene double bond across the 5,8-positions of the naphthalene ring. It is clearly a product arising directly from the exciplex and precludes the formation of separated radical ions, since a solvent-separated radical cation of 5 would invariably add methanol or alternatively undergo deprotonation prior to reaction with the arene radical anion. In fact, the formation of this cycloadduct is insensitive to solvent polarity and proceeds as efficiently in benzene (Reaction 14, Table 3) as it does in methanol or acetonitrile (Reaction 13, Table 3). It is surprising that addition occurs across the 5,8-positions rather than across the 1,4-positions. Most of the spin and charge densities are expected to reside at the cyano-substituted ring-carbon atoms and an exciplex of substantial charge transfer character would involve the allene complexing preferentially with the cyano-substituted ring. However, exciplex formation is known to be very sensitive to steric interactions and the unusual regiochemistry that is observed is most likely a consequence of steric inhibitions exerted by the cyano substituents.
Conclusions
The photoinduced nucleophile–olefin combination, aromatic substitution (photo-NOCAS) reaction has been successfully extended to include the reactions between aliphatic allenes and cyanoarenes. Although the yields are only moderate, the reactions mentioned represent simple and straightforward one-pot synthetic methods for the products shown, which, to the best of our knowledge, are all new compounds.More importantly, this study has provided us with further support for establishing an accurate mechanistic scheme for the photo-NOCAS reaction, which we have used in a lot of our previous work as a mechanistic framework for understanding the behaviour of photogenerated radical ions.1,3–5 The strict regiochemical control, involving the exclusive addition of the nucleophile to the central allenic carbon and the cyanoarene to the terminal one, supplements earlier studies from our laboratory with conjugated dienes1f and firmly establishes the sequence of mechanistic events in the photo-NOCAS reaction, with nucleophilic trapping of the olefin radical cation occurring prior to addition to the cyanoarene radical anion.
The drastic variations in the regiochemical selectivity observed in the reactions involving 1,1-dimethylallene (5) highlight the difficulty in predicting the preferred site of reactivity in non-symmetrical allylic radicals. The reaction outcome is most likely determined by a fine balance between two opposing factors: steric hindrance at the reaction site and spin density distribution in the allylic radical.
The reactions involving 1,4-dicyanonaphthalene (3) indicate that the photo-NOCAS reaction takes a different course with dinuclear cyanoaromatics, favouring addition of the olefin–nucleophile adduct radical to the cyanoarene over substitution, most probably a consequence of a lower rearomatisation energy. In these cases, the primary photochemical products were too photoreactive to be detected; the isolated materials resulted from intramolecular [2π + 2π] cycloaddition and nucleophile addition–cyclisation reactions of these elusive primary products.
Experimental
General information
Photochemical reactions were monitored and analysed by gas chromatography–mass spectrometry (GC–MS) using an HP 5890 gas chromatograph with an SPB-5 (Supelco) bonded 5% diphenylsiloxane–95% dimethylsiloxane fused silica WCOT column (25 m × 0.20 mm, 0.33 μm film thickness) and an HP 5970 mass selective detector. Quantitative gas chromatographic analysis was performed on a Perkin-Elmer AutoSystem XL gas chromatograph equipped with an autosampler, flame ionization detector (FID) and an MDN-5S (Supelco) bonded and crosslinked (5% phenyl) methylpolysiloxane fused silica WCOT column (30 m × 0.25 mm, 0.5 μm film thickness).Preparative separation of product mixtures was performed using flash chromatography on a 15 cm × 5 cm silica gel (Aldrich, 230–400 mesh, 60 Å) column.37 When necessary, this was followed by preparative, centrifugally accelerated, radial, thin-layer chromatography of the partially purified mixtures using a Chromatotron (Harrison Research) on 4, 2 or 1 mm silica gel (Aldrich, TLC grade 7749 with gypsum binder and fluorescent indicator) plates. The mobile phases were typically hexanes with increasing amounts of ethyl acetate. Collected fractions were analysed by TLC using silica gel plates (Aldrich, 250 μm plate thickness, 5–17 μm, 60 Å, with fluorescent indicator) and/or GC–MS.
All 1H and 13C NMR spectra were acquired on a Bruker AC 250F spectrometer at 250.13 MHz for 1H and 62.90 MHz for 13C. Chemical shifts are reported in ppm relative to tetramethylsilane (δ = 0 ppm) in 1H NMR spectra and chloroform-d (δ = 77.0 ppm) or acetonitrile-d3 (δ = 1.39 ppm) in 13C NMR spectra. Coupling constants (J values) are reported in Hz. The multiplicities of the decoupled 13C NMR signals were determined by J-Modulated Spin-Echo (J-MOD) experiments. Infrared spectra were recorded as films on sodium chloride plates on a Nicolet 510P FT-IR spectrophotometer and are reported in wavenumbers (cm−1). Melting points were determined using a Cybron Corporation Thermolyne apparatus equipped with a digital thermocouple (±0.1 °C) and are corrected. Elemental analysis was carried out by Canadian Microanalytical Service Ltd., Delta, BC. High-resolution mass spectrometry for exact mass determination was performed on a CEC 21-110 mass spectrometer using an electron-impact energy of 70 eV.
X-Ray crystallography![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) ‡
‡
Single crystal X-ray crystallographic structure determinations were performed at room temperature on a Rigaku AFC5R diffractometer equipped with a 12 kW rotating anode generator utilising graphite monochromated Cu-Kα (compounds 14, 16 and 19) or Mo-Kα (17, 18, 20 and 21) radiation. All data were corrected for Lorentz and polarization effects, while an empirical absorption correction (ψ scan; 14 and 17) and/or a correction for secondary extinction (14, 18 and 20) were applied as necessary. All calculations were formed using the teXsan crystallographic software package,38 except for the refinements of compounds 16 and 19 which were carried out using SHELXL-97.39 Problems with acquiring crystals of suitable size, shape and/or quality sometimes meant that not enough data could be collected for a full anisotropic refinement of the heavy atoms in a structure. In consequence, compounds 17 and 21 were refined totally isotropically, compounds 16, 19 and 20 were refined with some atoms anisotropic and other atoms isotropic, while compounds 14 and 18 were refined with all heavy atoms anisotropic. In all cases the reflection to parameter ratio was maintained at 5.0 or greater. Disorder was observed in only one structure (20) where the methoxy carbon (C34) was split over two positions, each with an occupancy of one half. In all structures, hydrogen atoms were placed in geometrically calculated positions and not refined.
Materials
1,4-Dicyanobenzene (98%, Aldrich) was purified by treatment with Norite in methylene chloride, followed by sublimation and recrystallisation from 95% ethanol. 1,4-Dicyanonaphthalene was prepared and purified as indicated previously.40 Biphenyl (99%, Aldrich) was recrystallised from methanol. 1,2,4,5-Tetracyanobenzene (Pfaltz and Bauer), tetramethylallene (97%, Aldrich), and 1,1-dimethylallene (98%, Aldrich) were used as received. Acetonitrile was distilled twice, first from sodium hydride and then from phosphorus pentaoxide. It was then passed through a column of basic alumina, refluxed over calcium hydride for 24 h under a nitrogen atmosphere, fractionally distilled and stored over 3 Å molecular sieves (Aldrich). Methanol was purified by reflux and distillation over magnesium and stored over 3 Å molecular sieves (Aldrich). Hexanes for preparative chromatography were distilled prior to use while ethyl acetate was used without further purification.Irradiations
Investigative irradiations were performed in 2 cm3 sample volumes in 20 cm × 0.5 cm tubes using a 1 kW medium-pressure mercury-arc lamp (CGE) fitted with a quartz water-cooled jacket immersed in a bath at 5 °C. Reactions used for quantitative analyses were performed in 10 cm3 sample volumes in 20 cm × 1 cm tubes while large-scale preparative photochemical reactions were carried out in 60–160 cm3 volumes in several 20 cm × 2 cm tubes. Tube distance from lamp axis was ca. 6 cm. All irradiations were carried out behind Pyrex (λ > 280 nm). Reaction details reported below are for the 10 cm3 quantitative-analysis samples. All yields were calibrated with respect to consumed cyanoarene. All quantitative-analysis GLC runs were done in triplicate and used an internal standard method for calibration. Pure samples of all products were isolated from large-scale photoreactions (60–160 cm3) identical in composition to the reactions reported below.4-(2,4,5-Tricyanophenyl)-3-methoxy-2,4-dimethylpent-2-ene (7). Colourless blocks, mp 166.9–167.6 °C (from hexanes) (Found: C, 72.9; H, 5.8; N, 15.1. C17H17N3O requires C, 73.1; H, 6.1; N, 15.0%); νmax(film; NaCl)/cm−1 3114 (m), 3042 (s), 2990 (s), 2970 (m), 2942 (s), 2916 (m), 2237 (s), 1492 (m), 1445 (m), 1371 (m), 1202 (m), 1193 (m), 1155 (m), 1131 (s), 1120 (s), 1077 (s), 936 (m) and 924 (s); δH(250.13 MHz; CDCl3; Me4Si) 1.00 (3 H, s), 1.62 (6 H, s), 1.73 (3 H, s), 3.73 (3 H, s), 7.94 (1 H, s) and 8.03 (1H, s); δC(62.90 MHz; CDCl3) 18.8 (q), 19.4 (q), 28.2 (q), 45.2 (s), 61.4 (q), 113.6 (s), 113.8 (s), 114.41 (s), 114.42 (s), 117.0 (s), 119.1 (s), 119.2 (s), 130.4 (d), 139.0 (d), 153.5 (s), 160.4 (s); m/z 279 (M˙+, 13%), 264 (100), 248 (86), 234 (81) and 194 (49).
1-(2,4,5-Tricyanophenyl)-2-methoxy-3-methylbut-2-ene (8). Pale yellow plates, mp 122.3–123.6 °C (from hexanes); νmax(film; NaCl)/cm−1 3111 (w), 3043 (m), 2993 (m), 2936 (s), 2831 (m), 2240 (s), 1682 (w), 1603 (m), 1489 (s), 1454 (m), 1386 (m), 1257 (m), 1196 (m), 1132 (s), 1016 (s) and 913 (m); δH(250.13 MHz; CDCl3; Me4Si) 1.75 (3 H, s), 1.76 (3 H, s), 3.48 (3 H, s), 3.83 (2 H, s), 7.82 (1 H, s) and 8.02 (1 H, s); δC(62.90 MHz; CDCl3) 17.2 (q), 19.1 (q), 32.2 (t), 57.6 (q), 113.7 (s), 114.2 (s), 114.5 (s), 114.7 (s), 117.8 (s), 119.3 (s), 121.1 (s), 134.9 (d), 136.7 (d), 144.0 (s) and 149.8 (s); m/z 251 (M˙+, 63%), 236 (76), 220 (100), 166 (86), 139 (61) and 85 (49) (M˙+, 251.1061. C15H13N3O requires M, 251.1058).
3-(2,4,5-Tricyanophenyl)-2-methoxy-3-methylbut-1-ene (9). Pale yellow plates, mp 129.6–130.8 °C (from hexanes); νmax(film; NaCl)/cm−1 3108 (w), 3041 (m), 2971 (m), 2944 (w), 2239 (m), 1609 (s), 1452 (m), 1370 (m), 1295 (m), 1279 (m), 1196 (m), 1175 (m), 1166 (m), 1148 (m), 1049 (s), 928 (s) and 811 (m); δH(250.13 MHz; CDCl3; Me4Si) 1.66 (6 H, s), 3.52 (3 H, s), 4.28 (1 H, d, J 3.6), 4.32 (1 H, d, J 3.6), 7.90 (1 H, s) and 8.01 (1 H, s); δC(62.90 MHz; CDCl3) 27.5 (q), 45.4 (s), 55.4 (q), 83.5 (t), 113.6 (s), 114.3 (s), 114.5 (s), 115.3 (s), 116.9 (s), 119.1 (s), 132.8 (d), 138.9 (d), 156.8 (s) and 165.4 (s); m/z 251 (M˙+, 29%), 236 (100), 220 (30), 204 (75) and 57 (79) (M˙+, 251.1076. C15H13N3O requires M, 251.1058).
4-(4-Cyanophenyl)-3-methoxy-2,4-dimethylpent-2-ene (10). Colourless oil; νmax(film; NaCl)/cm−1 2974 (m), 2934 (m), 2838 (w), 2227 (s), 1605 (m), 1503 (m), 1465 (m), 1449 (m), 1198 (m), 1152 (m), 1127 (s), 1108 (s), 1072 (s), 1020 (m), 992 (m) and 838 (m); δH(250.13 MHz; CDCl3; Me4Si) 1.01 (3 H, s), 1.48 (6 H, s), 1.68 (3 H, s), 3.66 (3 H, s) and 7.41–7.59 (4 H, m, AA′BB′); δC(62.90 MHz; CDCl3) 19.0 (q), 19.4 (q), 29.2 (q), 45.1 (s), 60.9 (q), 109.0 (s), 118.2 (s), 119.1 (s), 126.5 (d), 132.1 (d), 155.9 (q) and 156.7 (q); m/z 229 (M˙+, 67%), 214 (48), 197 (47), 182 (100), 116 (65) and 70 (48) (M˙+, 229.1467. C15H19NO requires M, 229.1457).
3-(4-Cyanophenyl)-2-methoxy-3-methylbut-1-ene (11). Colourless oil; νmax(film; NaCl)/cm−1 2975 (s), 2926 (s), 2852 (m), 2228 (s), 1606 (s), 1505 (s), 1465 (m), 1175 (m), 1095 (m) and 840 (m); δH(250.13 MHz; CDCl3; Me4Si) 1.46 (6 H, s), 3.47 (3 H, s), 4.09 (1 H, d, J 2.8), 4.21 (1 H, d, J 2.8) and 7.40–7.59 (4 H, m, AA′BB′); δC(62.90 MHz; CDCl3) 27.7 (q), 44.2 (s), 55.1 (q), 80.6 (t), 109.6 (s), 119.1 (s), 126.8 (d), 131.9 (d), 153.6 (s) and 168.9 (s); m/z 201 (M˙+, 29%), 186 (56), 169 (83), 154 (100) and 116 (87) (M˙+, 201.1159. C12H11N requires M, 201.1154).
1-(4-Cyanophenyl)-2-methoxy-3-methylbut-2-ene (12). m/z 201 (M˙+, 66%), 186 (26), 154 (55) and 116 (100).
3-(4-Cyanophenyl)-3-methylbut-1-yne (13). Colourless oil; νmax(film; NaCl)/cm−1 3297 (m), 2979 (s), 2933 (m), 2229 (s), 1607 (m), 1504 (m), 1457 (w), 1403 (w), 1364 (w), 1244 (w), 1096 (m), 1019 (w) and 839 (s); δH(250.13 MHz; CDCl3; Me4Si) 1.61 (6 H, s), 2.40 (1 H, s) and 7.60–7.70 (4 H, m, AA′BB′); δC(62.90 MHz; CDCl3) 31.2 (q), 36.2 (s), 70.9 (d), 89.4 (s), 110.5 (s), 118.9 (s), 126.4 (d), 132.2 (d) and 151.7 (s); m/z 169 (M˙+, 5%), 154 (100) and 127 (38) (M˙+, 169.0898. C12H11N requires M, 169.0891).
1,5-Dicyano-3-methoxy-2,2,4,4-tetramethyl-6,7-benzotricyclo[3.2.2.03,8]nonane (14). Colourless plates, mp >163 °C (decomp.) (from hexanes); νmax(film; NaCl)/cm−1 2991 (s), 2957 (s), 2837 (w), 2236 (m), 1489 (s), 1449 (s), 1392 (m), 1378 (m), 1264 (m), 1153 (s), 1145 (s), 1121 (m), 1095 (m), 1072 (s), 1050 (m), 1018 (m) and 758 (s); δH(250.13 MHz; CDCl3; Me4Si) 0.62 (3 H, s), 1.08 (3 H, s), 1.49 (3 H, s), 1.69 (3 H, s), 1.88 (1 H, d, J 12.8), 2.71 (1 H, dd, J 12.8, 4.6), 3.40 (3 H, s), 3.45 (1 H, d, J 4.6) and 7.75–8.55 (4 H, m); δC(62.90 MHz; CDCl3) 22.0 (q), 23.5 (q), 26.0 (q), 27.0 (q), 34.7 (t), 42.3 (s), 47.3 (d), 54.79 (s), 54.82 (s), 55.1 (q), 57.0 (s), 89.1 (s), 119.6 (s), 119.8 (s), 127.3 (d), 128.1 (d), 128.7 (d), 129.1 (d), 130.4 (s) and 135.7 (s); m/z 306 (M˙+, 0.6%), 259 (12), 238 (41), 127 (89), 95 (86) and 69 (100).
Crystal data: C20H22N2O, M = 306.41, monoclinic, a = 9.927(1), b = 11.116(1), c = 15.3609(7) Å, β = 103.868(5)°, V = 1645.6(2) Å3, T = 296 K, space group P21/n (no. 14), Z = 4, μ(Cu-Kα) = 6 cm−1, 3010 reflections measured, 2831 unique (Rint = 0.031). The final R and Rw were 0.041 and 0.039 respectively and are based on 1432 observed reflections (I > 3.00σ(I)) and 209 parameters.
cis-1,4-Dicyano-6,6,8,8-tetramethyl-7-oxo-2,3-benzo-cis-bicyclo[3.3.1]nonane (16). Colourless needles, mp 252.9–253.8 °C (from methanol); νmax(film; NaCl)/cm−1 2970 (m), 2964 (m), 2904 (m), 2239 (m), 1701 (vs), 1491 (m), 1477 (m), 1464 (m), 1447 (m), 1390 (m), 1371 (m), 1043 (m) and 767 (s); δH(250.13 MHz; CDCl3; Me4Si) 1.05 (3 H, s), 1.14 (3 H, s), 1.46 (3 H, s), 1.62 (3 H, s), 2.61 (1 H, br s), 2.82 (1 H, dd, J 14.5, 2.4), 3.26 (1 H, ddd, J 14.5, 4.0, 1.5), 4.19 (1 H, br s) and 7.26–7.60 (4 H, m); δC(62.90 MHz; CDCl3) 23.6 (q), 24.5 (q), 26.9 (t), 30.2 (q), 33.1 (d), 43.7 (d), 46.5 (s), 46.7 (s), 51.4 (s), 120.2 (s), 120.6 (s), 125.9 (s), 128.6 (d), 130.2 (d), 130.3 (d), 131.4 (d), 131.7 (s) and 213.5 (s); m/z 292 (M˙+, 2%), 113 (100) and 95 (25).
Crystal data: C19H20N2O, M = 292.38, monoclinic, a = 6.227(1), b = 16.056(1), c = 15.917(1) Å, β = 92.21(1)°, V = 1590.3(3) Å3, T = 296 K, space group Cc (no. 9), Z = 4, μ(Cu-Kα) = 6 cm−1, 1482 reflections measured, 1421 unique (Rint = 0.029). The final R1 and wR2 were 0.049 and 0.168 respectively and are based on 808 observed reflections (I > 3.00σ(I)) and 139 parameters.
1,5-Dicyano-3-methoxy-4,4-dimethyl-6,7-benzotricyclo[3.2.2.03,8]nonane (17). Colourless plates, mp 126.4–127.2 °C (from hexanes–ethyl acetate); νmax(film; NaCl)/cm−1 2981 (s), 2943 (s), 2832 (w), 2238 (m), 1489 (m), 1452 (s), 1278 (m), 1155 (s), 1115 (s), 1106 (m), 1088 (m) and 1073 (s); δH(250.13 MHz; CDCl3; Me4Si) 0.83 (3 H, s), 1.43 (3 H, s), 1.82 (1 H, d, J 12.8), 2.23 (1 H, d, J 12.2), 2.71 (1 H, dd, J 12.8, 4.5), 3.12 (1 H, d, J 12.2), 3.27 (3 H, s), 3.51 (1 H, d, J 4.5) and 7.38–7.70 (4 H, m); δC(62.90 MHz; CD3CN) 22.3 (q), 23.7 (q), 31.2 (s), 35.6 (t), 42.1 (t), 50.5 (d), 52.7 (q), 52.9 (s), 55.9 (s), 84.8 (s), 120.8 (s), 122.3 (s), 127.5 (d), 127.6 (d), 129.0 (d), 130.6 (d), 133.1 (s) and 136.7 (s); m/z 278 (M˙+, 2%), 152 (9), 99 (93) and 67 (100) (M˙+, 278.1416. C18H18N2O requires M, 278.1419).
Crystal data: C72H72N8O4 [4 (C18H18N2O)], M = 1113.41, orthorhombic, a = 14.294(5), b = 16.505(6), c = 25.334(5) Å, V = 5977(3) Å3, T = 296 K, space group Pca21 (no. 29), Z = 4, μ(Mo-Kα) = 0.78 cm−1, 6310 reflections measured. The final R and Rw were 0.082 and 0.080 respectively and are based on 1679 observed reflections (I > 3.00σ(I)) and 336 parameters.
2,5-Dicyano-1-methoxy-9,9-dimethyl-3,4-benzotricyclo[3.3.1.02,7]nonane (18). Colourless blocks, mp 148.7–149.8 °C (from hexanes); νmax(film; NaCl)/cm−1 2976 (s), 2940 (m), 2838 (w), 2240 (m), 1486 (s), 1455 (s), 1392 (w), 1373 (w), 1276 (m), 1236 (s), 1220 (s), 1208 (m), 1151 (s), 1131 (s), 1072 (m), 1057 (s), 1026 (m) and 760 (s); δH(250.13 MHz; CDCl3; Me4Si) 0.43 (3 H, s), 1.43 (3 H, s), 1.71 (1 H, ddd, J 12.8, 7.2, 1.2), 2.20 (1 H, d, J 10.7), 2.50 (1 H, d, J 12.8), 2.62 (1 H, t, J 7.2), 2.99 (1 H, ddd, J 10.7, 7.2, 1.2), 3.54 (3 H, s) and 7.27–7.65 (4 H, m); δC(62.90 MHz; CDCl3) 21.2 (q), 21.3 (q), 32.2 (d), 34.1 (t), 34.2 (t), 42.3 (s), 48.3 (s), 49.6 (s), 54.5 (d), 84.6 (s), 118.4 (s), 119.6 (s), 123.1 (d), 125.2 (d), 128.2 (s), 128.4 (d), 128.7 (d) and 136.7 (s); m/z 278 (M˙+, 4%), 237 (11), 178 (22), 99 (80) and 67 (100).
Crystal data: C18H18N2O, M = 278.35, orthorhombic, a = 12.214(2), b = 13.305(3), c = 9.083(2) Å, V = 1476.0(4) Å3, T = 296 K, space group P212121 (no. 19), Z = 4, μ(Mo-Kα) = 0.8 cm−1, 2481 reflections measured. The final R and Rw were 0.040 and 0.043 respectively and are based on 1071 observed reflections (I > 3.00σ(I)) and 191 parameters.
cis-1,4-Dicyano-7,7-dimethoxy-6,6-dimethyl-2,3-benzo-cis-bicyclo[3.3.1]nonane (19). Colourless blocks, mp 167.5–168.4 °C (from methanol); νmax(film; NaCl)/cm−1 2990 (m), 2967 (m), 2951 (m), 2835 (m), 2237 (m), 1466 (m), 1448 (m), 1137 (s), 1125 (s), 1112 (s), 1054 (s), 1038 (m), 1030 (m), 979 (m) and 910 (m); δH(250.13 MHz; CDCl3; Me4Si) 1.23 (3 H, s), 1.29 (3 H, s), 2.13 (3H, s), 2.18–2.31 (3 H, m), 2.37 (1 H, dt, J 13.4, 5.2), 2.62 (1 H, ddd, J 13.4, 3.7, 0.9), 3.14 (3 H, s), 4.30 (1 H, s) and 7.27–7.32 (4 H, m); δC(62.90 MHz; CDCl3) 23.6 (q), 25.0 (q), 29.5 (t), 31.4 (d), 37.0 (s), 37.2 (t), 42.5 (s), 46.6 (d), 47.4 (q), 50.1 (q), 100.1 (s), 122.0 (s), 122.4 (s), 127.0 (d), 127.9 (d), 128.7 (d), 129.0 (d), 130.8 (s) and 133.9 (s); m/z 310 (M˙+, 8%), 279 (18), 267 (15), 180 (33), 99 (77), 88 (100) and 67 (31).
Crystal data: C19H22N2O2, M = 310.39, monoclinic, a = 13.917(1), b = 8.5864(6), c = 14.304(1) Å, β = 105.596(7)°, V = 1646.3(2) Å3, T = 296 K, space group P21/n (no. 14), Z = 4, μ(Cu-Kα) = 6.5 cm−1, 1165 reflections measured, 989 unique (Rint = 0.024). The final R1 and wR2 were 0.058 and 0.184 respectively and are based on 860 observed reflections (I > 3.00σ(I)) and 113 parameters.
trans-1,4-Dicyano-7,7-dimethoxy-8,8-dimethyl-2,3-benzo-cis-bicyclo[3.3.1]nonane (20). Colourless blocks, mp 122.9–124.6 °C (from hexanes); νmax(film; NaCl)/cm−1 2990 (m), 2949 (m), 2834 (m), 2239 (m), 1492 (m), 1449 (m), 1171 (m), 1129 (s), 1122 (s), 1096 (m), 1073 (m), 1053 (s), 1044 (s) and 972 (m); δH(250.13 MHz; CDCl3; Me4Si) 1.23 (3 H, s), 1.42 (3 H, s), 1.99 (1 H, dd, J 15.0, 4.9), 2.14 (1 H, ddd, J 14.7, 4.0, 1.8), 2.46 (1 H, dt, J 14.7, 2.0), 2.70 (3 H, s), 2.71–2.77 (2 H, m), 3.21 (3 H, s), 4.37 (1 H, d, J 6.1) and 7.25–7.65 (4 H, m); δC(62.90 MHz; CDCl3) 20.3 (q), 24.1 (q), 29.3 (d), 31.3 (t), 32.2 (t), 37.1 (d), 47.15 (s), 47.17 (s), 49.9 (q), 50.0 (q), 100.7 (s), 120.2 (s), 122.3 (s), 126.3 (d), 127.0 (d), 128.6 (d), 129.5 (d), 131.1 (s) and 133.5 (s); m/z 310 (M˙+, 33%), 279 (100), 267 (94) and 88 (56).
Crystal data: C38H44N4O4 [2 (C19H22N2O2)], M = 620.78, monoclinic, a = 16.452(3), b = 9.016(2), c = 23.003(2) Å, β = 95.35(1)°, V = 3397.3(9) Å3, T = 296 K, space group P21/a (no. 14), Z = 4, μ(Mo-Kα) = 0.8 cm−1, 2987 reflections measured, 2685 unique (Rint = 0.063). The final R and Rw were 0.059 and 0.055 respectively and are based on 1622 observed reflections (I > 3.00σ(I)) and 300 parameters.
7-Isopropylidene-5,6-(1′,4′-dicyanobenzo)bicyclo[2.2.2]oct-2-ene (21). Colourless blocks, mp 178.1–178.9 °C (from hexanes); νmax(film; NaCl)/cm−1 3081 (w), 3066 (w), 2978 (m), 2915 (s), 2855 (m), 2233 (s), 1474 (m), 1436 (m), 1400 (s), 1374 (m), 1335 (s), 1251 (m), 1175 (m), 1158 (m), 1123 (m) and 826 (s); δH(250.13 MHz; CDCl3; Me4Si) 1.55 (3 H, s), 1.91 (3 H, s), 2.04 (1 H, d, J 15.3), 2.30 (1 H, d, J 15.3), 4.54 (1 H, m), 5.24 (1 H, dd, J 5.5, 1.8), 6.61 (2 H, m) and 7.44 (2 H, s); δC(62.90 MHz; CDCl3) 20.2 (q), 21.7 (q), 32.1 (t), 39.9 (d), 43.5 (d), 110.3 (s), 110.8 (s), 116.29 (s), 116.32 (s), 125.4 (s), 126.0 (s), 128.3 (d), 128.5 (d), 133.4 (d), 134.6 (d), 148.5 (s) and 149.2 (s); m/z 246 (M˙+, 82%), 231 (100), 178 (22) and 68 (72).
Crystal data: C17H14N2, M = 246.31, orthorhombic, a = 13.37(1), b = 25.49(1), c = 7.920(7) Å, V = 2698(3) Å3, T = 296 K, space group Pbca (no. 61), Z = 8, μ(Mo-Kα) = 0.7 cm−1, 2657 reflections measured. The final R and Rw were 0.083 and 0.087 respectively and are based on 460 observed reflections (I > 3.00σ(I)) and 77 parameters.
Acknowledgements
This work was supported financially by the National Sciences and Engineering Research Council (NSERC) of Canada. D. Mangion is grateful for a scholarship from the Izaak Walton Killam Memorial Foundation. We would like to thank Dr D. L. Hooper at the Atlantic Regional Magnetic Resonance Center at Dalhousie University for help with the NMR spectroscopy and with the preparation of the manuscript, Dr J.-H. Kim for the exact mass measurements and Ms N. Mora-Diez for the AM1 calculations. We are also thankful to Dr R. M. Borg at the University of Malta for sharing some of the preliminary results that encouraged us to do this work.References
- (a) R. M. Borg, D. R. Arnold and T. S. Cameron, Can. J. Chem., 1984, 62, 1785 CAS; (b) D. R. Arnold and X. Du, J. Am. Chem. Soc., 1989, 111, 7666 CrossRef CAS; (c) K. McMahon and D. R. Arnold, Can. J. Chem., 1993, 71, 450 CAS; (d) D. R. Arnold and X. Du, Can. J. Chem., 1994, 72, 403 CAS; (e) D. R. Arnold, K. A. McManus and X. Du, Can. J. Chem., 1994, 72, 415 CAS; (f) K. A. McManus and D. R. Arnold, Can. J. Chem., 1994, 72, 2291 CAS; (g) D. R. Arnold, X. Du and H. J. P. de Lijser, Can. J. Chem., 1995, 73, 522 CAS; (h) D. A. Connor, D. R. Arnold, P. K. Bakshi and T. S. Cameron, Can. J. Chem., 1995, 73, 762 CAS; (i) K. A. McManus and D. R. Arnold, Can. J. Chem., 1995, 73, 2158 CAS; (j) D. R. Arnold, D. A. Connor, K. A. McManus, P. K. Bakshi and T. S. Cameron, Can. J. Chem., 1996, 74, 602 CAS; (k) H. J. P. de Lijser and D. R. Arnold, Can. J. Chem., 1997, 62, 8432 CAS; (l) D. R. Arnold and K. A. McManus, Can. J. Chem., 1998, 76, 1238 CrossRef CAS.
- (a) J. L. Stavinoha and P. S. Mariano, J. Am. Chem. Soc., 1981, 103, 3136 CrossRef CAS; (b) H. Weng and H. D. Roth, J. Org. Chem., 1995, 60, 4136 CrossRef CAS; (c) R. Torriani, M. Mella, E. Fasani and A. Albini, Tetrahedron, 1997, 33, 2573 CrossRef CAS; (d) H. Weng, C. Scarlata and H. D. Roth, J. Am. Chem. Soc., 1996, 118, 10947 CrossRef CAS; (e) T. Herbertz and H. D. Roth, J. Am. Chem. Soc., 1996, 118, 10954 CrossRef CAS; (f) T. Herbertz, F. Blume and H. D. Roth, J. Am. Chem. Soc., 1998, 120, 4591 CrossRef CAS.
- D. R. Arnold and M. S. Snow, Can. J. Chem., 1988, 66, 3012 CAS.
- (a) D. R. Arnold, M. S. W. Chan and K. A. McManus, Can. J. Chem., 1996, 74, 2143 CAS; (b) M. S. W. Chan and D. R. Arnold, Can. J. Chem., 1997, 75, 1810 CAS.
- D. R. Arnold. K. A. McManus and M. S. W. Chan, Can. J. Chem., 1997, 75, 1055.
- (a) K. Somekawa, K. Haddaway, P. S. Mariano and J. A. Tossell, J. Am. Chem. Soc., 1984, 106, 3060 CrossRef CAS; (b) K. Haddaway, K. Somekawa, P. Fleming, J. A. Tossell and P. S. Mariano, J. Org. Chem., 1987, 52, 4239 CrossRef CAS.
- (a) M. W. Klett and R. P. Johnson, Tetrahedron Lett., 1983, 24, 1107 CrossRef CAS; (b) M. W. Klett and R. P. Johnson, J. Am. Chem. Soc., 1985, 107, 6615 CrossRef CAS.
- (a) R. A. Neunteufel and D. R. Arnold, J. Am. Chem. Soc., 1973, 95, 4080 CrossRef CAS; (b) A. J. Maroulis and D. R. Arnold, Synthesis, 1979, 10, 819 CrossRef; (c) D. R. Arnold, X. Du and J. Chen, Can. J. Chem., 1995, 73, 307 CAS; (d) D. Mangion and D. R. Arnold, Can. J. Chem., 1999, 77, 1655 CrossRef CAS.
- (a) D. R. Arnold and A. H. Glick, J. Chem. Soc., Chem. Commun., 1966, 813 RSC; (b) H. Gotthardt, R. Steinmetz and G. S. Hammond, J. Org. Chem., 1968, 33, 2774 CAS; (c) H. J. T. Bos, C. Slagt and J. S. M. Boleij, Recl. Trav. Chim. Pays-Bas, 1970, 89, 1170 Search PubMed; (d) N. Ishibe and I. Tanigushi, Tetrahedron, 1971, 27, 4883 CrossRef CAS; (e) J. S. M. Boleij and H. J. T. Bos, Tetrahedron Lett., 1971, 3201 CrossRef CAS; (f) N. Ishibe, K. Hoshimoto and Y. Yamaguchi, J. Chem. Soc., Perkin Trans. 1, 1975, 318 RSC; (g) K. Ogino, T. Matsumoto, T. Kawai and S. Kozuka, J. Org. Chem., 1979, 44, 3352 CrossRef CAS; (h) W. R. McKay, J. Ounsworth, P. E. Sum and L. Weiler, Can. J. Chem., 1982, 60, 872 CAS; (i) D. Becker, M. Nagler, Z. Harel and A. Gillon, J. Org. Chem., 1983, 48, 2584 CrossRef CAS; (j) J. Kamphuis, P. D. J. Grootenhuis, A. P. Ruijter, R. G. Visser and H. J. T. Bos, Isr. J. Chem., 1985, 26, 120 Search PubMed; (k) L. K. Sydnes and W. Stensen, Acta Chem. Scand., Ser. B, 1986, 40, 657 Search PubMed.
- K. Maruyama and H. Imahori, J. Org. Chem., 1989, 54, 2692 CrossRef CAS.
- N. J. Turro and V. Ramamurthy, Tetrahedron Lett., 1976, 2423 CrossRef CAS.
- J. Kamphuis, H. J. T. Bos, R. J. Visser, B. H. Huizer and C. A. G. O. Varma, J. Chem. Soc., Perkin Trans. 2, 1986, 1867 RSC.
- Typically, completion of the photosensitisation cycle requires the sensitiser radical anion to reduce a radical species derived from the donor radical cation after nucleophilic trapping, deprotonation, desilylation or some other process. The reduction potentials of most typical radicals are too high for exergonic electron transfer with 1−−˙. (a) D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132 CrossRef CAS; (b) B. A. Sim, P. H. Milne, D. Griller and D. D. M. Wayner, J. Am. Chem. Soc., 1990, 112, 6635 CrossRef CASThe only cases in which photosensitisation of 1 is indeed observed involve the regeneration of 1 from 1−−˙via an electron transfer process with a carbocationic moiety. (c) T. Asanuma, M. Yamamoto and Y. Nishijima, J. Chem. Soc., Chem. Commun., 1975, 608 RSC; (d) H. Ikeda, T. Minegishi and T. Miyashi, J. Chem. Soc., Chem. Commun., 1994, 297 RSC.
- (a) M. Vanossi, M. Mella and A. Albini, J. Am. Chem. Soc., 1994, 116, 10070 CrossRef CAS; (b) M. Mella, M. Freccero and A. Albini, J. Org. Chem., 1994, 59, 1047 CrossRef CAS; (c) M. Mella, M. Fagnoni and A. Albini, J. Org. Chem., 1994, 59, 5614 CrossRef CAS; (d) E. Fasani, D. Peverali and A. Albini, Tetrahedron Lett., 1994, 35, 9275 CrossRef CAS; (e) M. Fagnoni, M. Mella and A. Albini, Tetrahedron, 1994, 50, 6401 CrossRef CAS; (f) Tetrahedron, 1995, 51, 859; Search PubMed; (g) M. Mella, M. Freccero and A. Albini, J. Chem. Soc., Chem. Commun., 1995, 41 RSC; (h) M. Fagnoni, M. Mella and A. Albini, J. Am. Chem. Soc., 1995, 117, 7877 CrossRef CAS; (i) M. Fagnoni, M. Vanossi, M. Mella and A. Albini, Tetrahedron, 1996, 52, 1785 CrossRef CAS; (j) M. Mella, M. Freccero, T. Soldi, E. Fasani and A. Albini, J. Org. Chem., 1996, 61, 1413 CrossRef CAS; (k) M. Mella, M. Fagnoni, M. Freccero, E. Fasani and A. Albini, Chem. Soc. Rev., 1998, 27, 81 RSC.
- S. L. Mattes and S. Farid , in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1983, vol. 6, p. 233. Search PubMed.
- G. Ginzburg, B. Zinger and J. Y. Becker, J. Electroanal. Chem., 1983, 57 CrossRef CAS.
- (a) R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706 CrossRef CAS; (b) R. S. Nicholson and I. Shain, Anal. Chem., 1965, 37, 178 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 Search PubMed.
- For some representative examples see: (a) T. Majima, C. Pac and H. Sakurai, J. Chem. Soc., Perkin Trans. 1, 1980, 2705 RSC; (b) D. S. Steichen and C. S. Foote, J. Am. Chem. Soc., 1981, 103, 1855 CrossRef CAS; (c) A. P. Schaap, S. Siddiqui, S. D. Gagnon and L. Lopez, J. Am. Chem. Soc., 1983, 105, 5149 CrossRef CAS; (d) S. C. Shim and J. S. Song, J. Org. Chem., 1986, 51, 2817 CrossRef CAS; (e) Y. Kuriyama, T. Arai, H. Sakuragi and K. Tokumaru, Chem. Lett., 1992, 879 CAS.
- (a) I. R. Gould, D. Ege, J. E. Moser and S. Farid, J. Am. Chem. Soc., 1990, 112, 4290 CrossRef CAS; (b) F. D. Lewis, A. M. Bedell, R. E. Dykstra, J. E. Elbert, I. R. Gould and S. Farid, J. Am. Chem. Soc., 1990, 112, 8055 CrossRef CAS.
- (a) C. Pac, A. Nakasone and H. Sakurai, J. Am. Chem. Soc., 1977, 99, 5806 CrossRef CAS; (b) T. Majima, C. Pac and H. Sakurai, J. Chem. Soc., Perkin Trans. 1, 1980, 2705 RSC; (c) T. Majima, C. Pac, A. Nakasone and H. Sakurai, J. Am. Chem. Soc., 1981, 103, 4499 CrossRef CAS; (d) S. L. Mattes, H. R. Luss and S. Farid, J. Phys. Chem., 1983, 87, 4779 CrossRef CAS; (e) T. Tamai, K. Mizuno, I. Hashida and Y. Otsuji, J. Org. Chem., 1992, 57, 5338 CrossRef CAS; (f) T. Yamashita, M. Yasuda, T. Isami, S. Nakano, K. Tanabe and K. Shima, Tetrahedron Lett., 1993, 34, 5131 CrossRef CAS.
- Similar increases in reaction efficiencies have also been reported with hydrocarbon co-donors (e.g., benzene) which possess too high oxidation potentials for exergonic electron transfer with the acceptor substrate (endergonic first step in Scheme 5). This is attributed to the formation of a π-complex between the aromatic hydrocarbon co-donor and the donor radical cation (generated in the PET step with the electron acceptor); this interaction stabilises the donor radical cation, enhancing its diffusion out of cage with the acceptor radical anion, thus suppressing BET; (a) K. Mizuno, K. Nakanishi and Y. Otsuji, Chem. Lett., 1988, 1833 CAS; (b) M. Yasuda, T. Isami, J. Kubo, M. Mizutani, T. Yamashita and K. Shima, J. Org. Chem., 1992, 57, 1351 CrossRef CAS; (c) K. Nakanishi, K. Mizuno and Y. Otsuji, Bull. Chem. Soc. Jpn., 1993, 66, 2371 CAS; (d) K. Mizuno, T. Tamai, T. Nishiyama, K. Tani, M. Sawasaki and Y. Otsuji, Angew. Chem., Int. Ed. Engl., 1994, 33, 2113 CrossRef.
- (a) J. J. McCullough, R. C. Miller and W. S. Wu, Can. J. Chem., 1977, 55, 2909 CAS; (b) D. R. Arnold, P. C. Wong, A. J. Maroulis and T. S. Cameron, Pure Appl. Chem., 1980, 52, 2609 CAS.
- F. D. Lewis and R. J. DeVoe, Tetrahedron, 1982, 38, 1069 CrossRef CAS.
- P. H. Mazzocchi and G. Fritz, J. Am. Chem. Soc., 1986, 108, 5362 CrossRef CAS.
- (a) P. S. Engel, D. J. Bishop and M. A. Page, J. Am. Chem. Soc., 1978, 100, 7009 CrossRef CAS; (b) D. L. Baulch, P. K. Chown and D. C. Montague, Int. J. Chem. Kinet., 1979, 11, 1055 CrossRef CAS.
-
E
½
ox was determined by means of an empirical correlation relating the peak oxidation potential (Eox, solution) to the ionisation potential (IP, gas phase): Eox = (0. 827 × IP − 5.40) V vs. Ag/Ag+;28Eox was corrected to E½ox by subtracting 0.03 V;17 referenced to SCE by adding 0.34 V. IP for (Z)-2-methoxybut-2-ene is 8.25 V.29.
- W. C. Neikam, G. R. Dimeler and M. M. Desmond, J. Electrochem. Soc., 1964, 111, 1190 CAS.
- H. Friege and M. Klessinger, J. Chem. Res. (S), 1977, 208 Search PubMed.
- K. Kokubo, T. Masaki and T. Oshima, Org. Lett., 2000, 2, 1979 CrossRef CAS.
- (a) F. D. Lewis, T. I. Ho and J. T. Simpson, J. Am. Chem. Soc., 1982, 104, 1924 CrossRef CAS; (b) F. D. Lewis, Acc. Chem. Res., 1986, 19, 401 CrossRef CAS.
- (a) D. R. Arnold and S. A. Mines, Can. J. Chem., 1987, 65, 2312 CAS; (b) D. R. Arnold and S. A. Mines, Can. J. Chem., 1989, 67, 689 CAS.
- (a) M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870 CrossRef CAS; (b) E. Breitmaier , Structure elucidation by NMR in organic chemistry, Wiley, Chichester, UK, 1993, p. 43. Search PubMed.
- H. Friebolin , Basic one- and two-dimensional NMR spectroscopy, VCH, New York, 1993, p. 91. Search PubMed.
- (a) A. Albini and S. Spreti, Tetrahedron, 1984, 40, 2975 CrossRef CAS; (b) Y. Kubo, K. Kiuchi and I. Inamura, Bull. Chem. Soc. Jpn., 1999, 72, 1101 CrossRef CAS.
- G. Le Guillanton, Q. T. Do and J. Simonet, Bull. Soc. Chim. Fr., 1990, 127, 427 Search PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- Molecular Structure Corporation, teXsan for Windows, version 1.5, Crystal Structure Analysis Package, 1997–8. Search PubMed.
- G. M. Sheldrick , SHELX-97, Program for Crystal Structure Determinations, 1997. Search PubMed.
- A. Okamoto, M. S. Snow and D. R. Arnold, Tetrahedron, 1986, 42, 6175 CrossRef CAS.
Footnotes |
| † The IUPAC names for the parent hydrocarbons that form the basis of structures 14–21 are tetracyclo[8.2.1.02,7.08,11]trideca-2(7),3,5-triene (14, 17), tricyclo[7.3.1.02,7]trideca-2(7),3,5-triene (15, 16, 19, 20), tetracyclo[7.3.1.02,11.03,8]trideca-3(8),4,6-triene (18) and tricyclo[6.2.2.02,7]dodeca-2,4,6-triene (21). |
| ‡ CCDC reference number 188/278. See http://www.rsc.org/suppdata/p2/b0/b007205m/ for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |